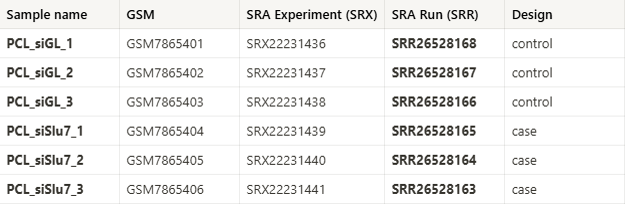
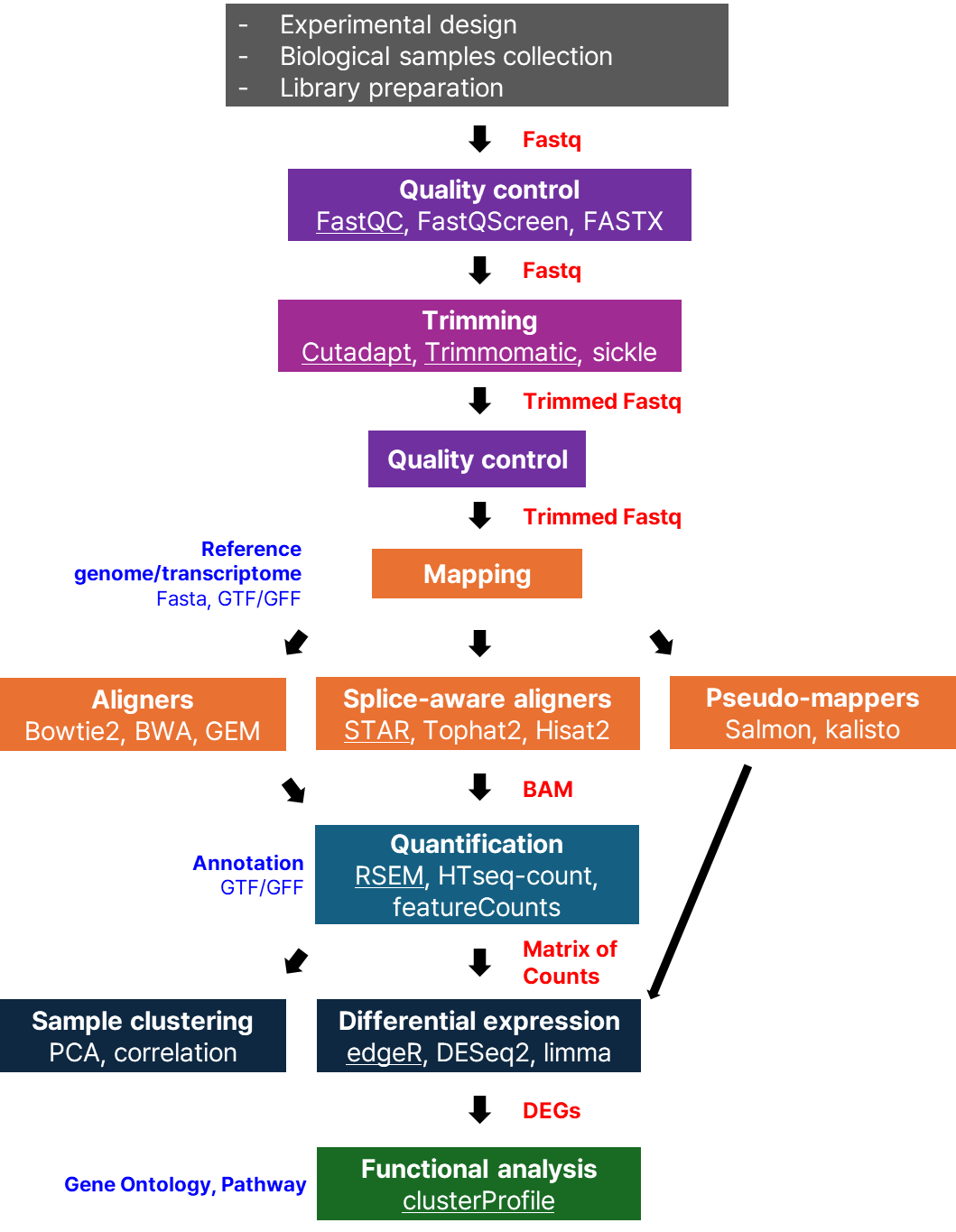
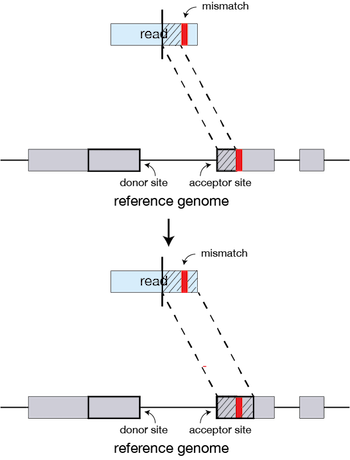
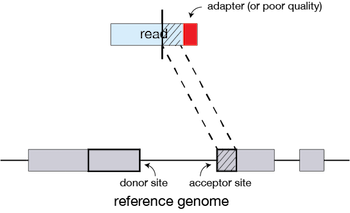
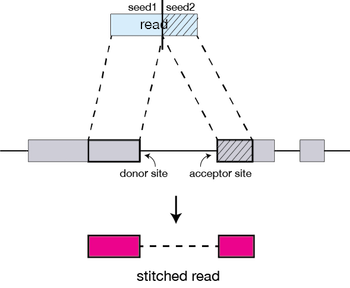
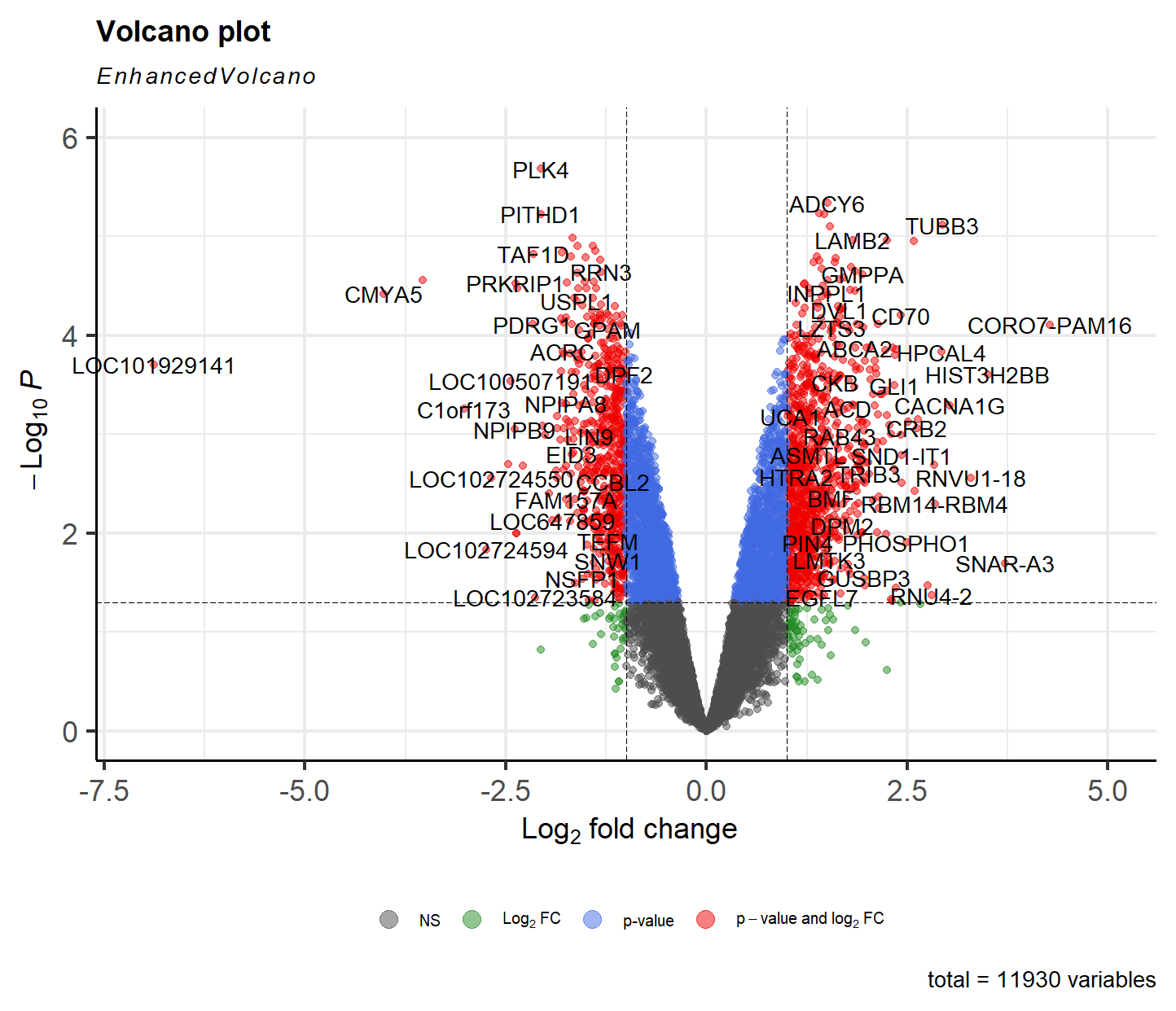
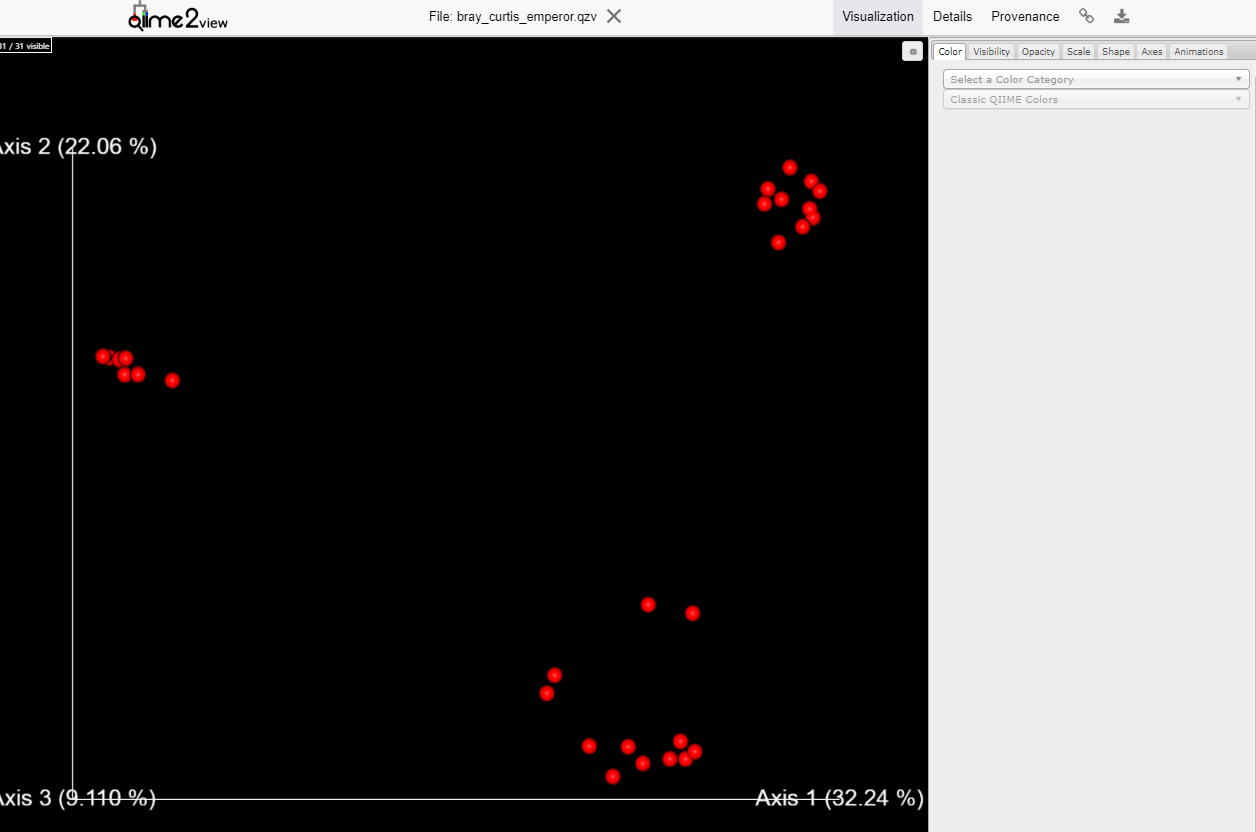
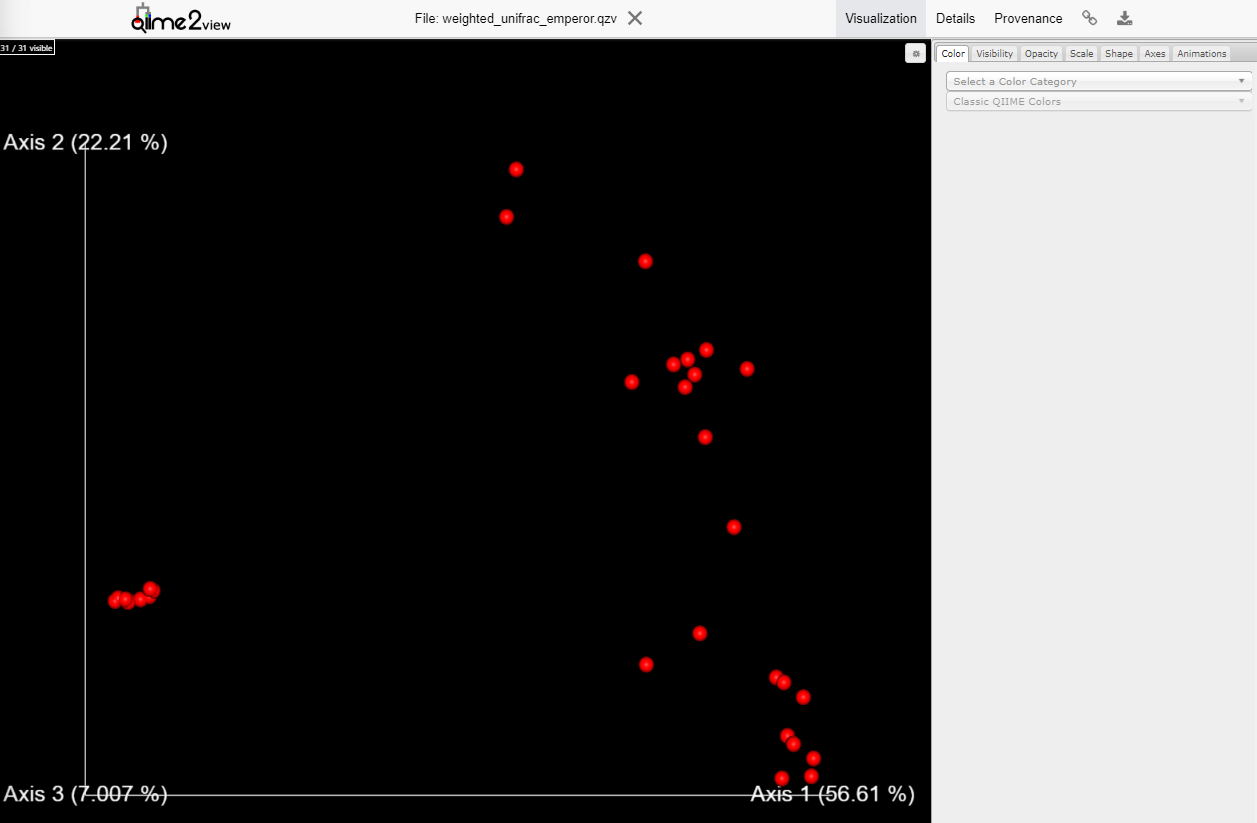
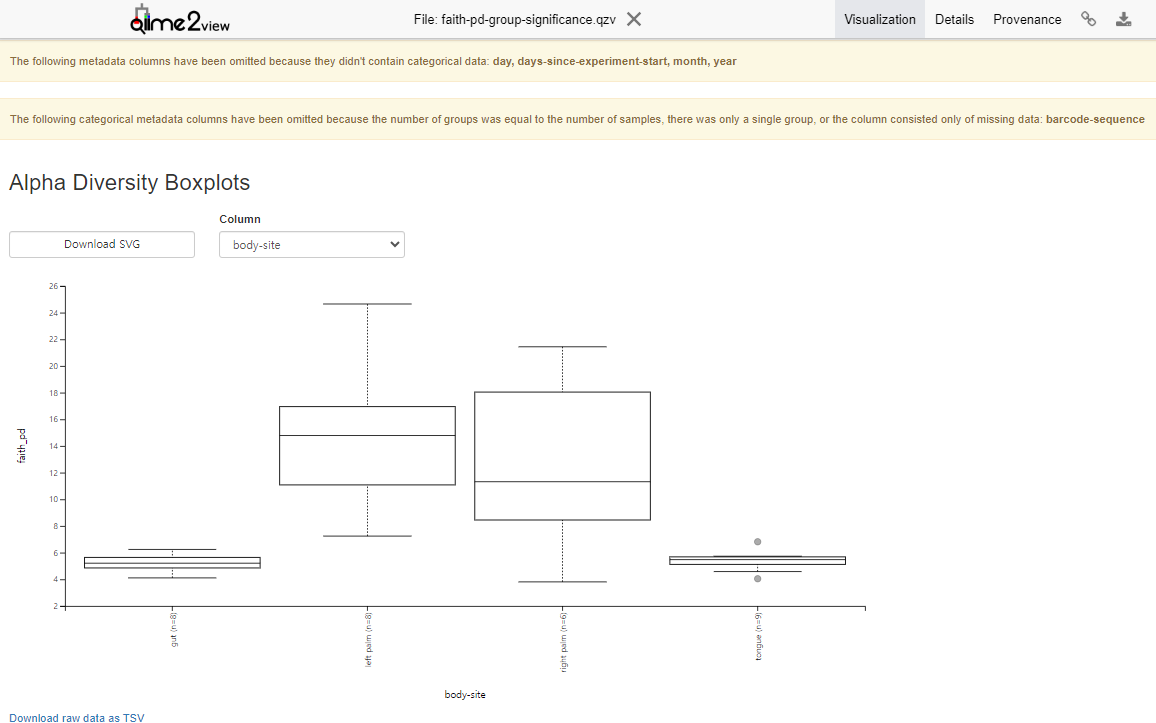
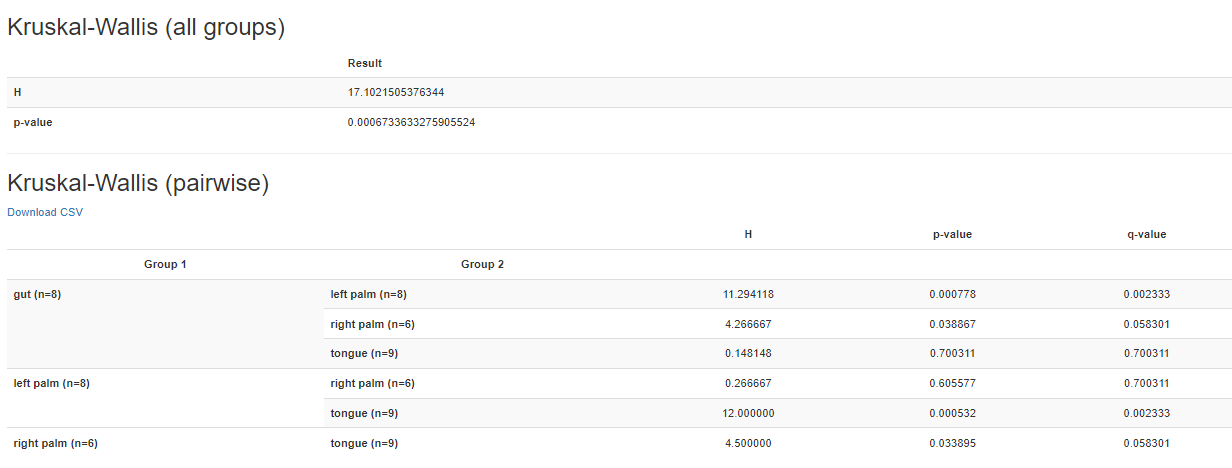
Variant Calling (From reads to short variants)
00. Introduction
전장 유전체 시퀀싱(WGS)은 유전체 전체를 분석하는 포괄적인 방법으로, 해당 튜토리얼에서는 Human 샘플에 대한 WGS 데이터에서 germline 변이 (SNP 및 INDEL)을 발굴하는 과정을 수행합니다.
본 튜토리얼에서 사용할 데이터셋은 미국의 국립표준기술연구소(NIST)의 Genome in a Bottle (GIAB) project 에 포함된 Ashkenazim Trio (아슈케나지 유대인) 데이터입니다.
샘플의 구성은 Son:HG002, Father:HG003 , Mother:HG004로 확인됩니다.
다운로드 링크 : https://github.com/genome-in-a-bottle/giab_data_indexes


01. QC (Quality control)
NGS는 기술적 한계와 실험적 원인에 의한 다양한 오류(error)의 가능성이 있습니다.
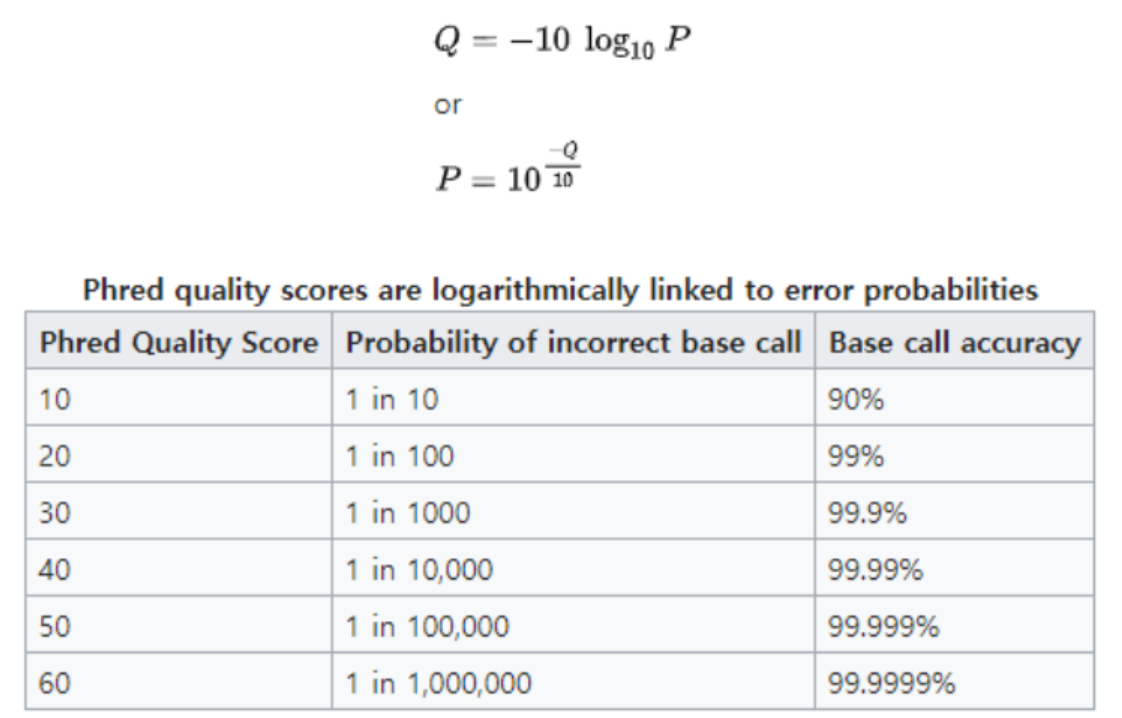
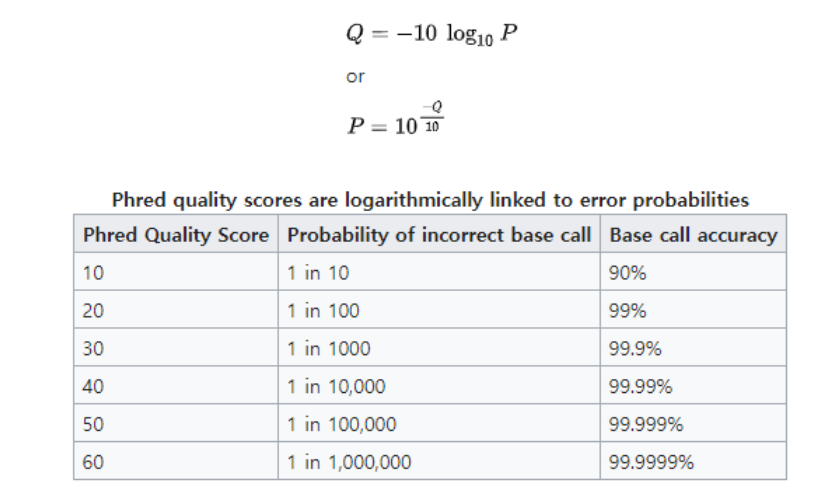
NGS 염기서열 분석 결과의 원시 데이터(raw data)에서는 각 base별로 추정 오류 확률을 수치로 나타내며 이를 Phred score라고 불리며 이는 각 염기의 품질을 나타내는 지표로 활용됩니다.
Phred score 20점은 해당 염기서열 결과가 오류일 확률이 1%이라는 것을 의미하며, 30점은 0.1%의 오류 확률을 의미합니다. 일반적으로 Q30 이상의 Phred 점수를 보이는 염기는 시퀀싱 품질이 우수하다고 판단하여 분석에 활용할 수 있습니다.
소개
Phred quality score는 NGS 시퀀싱으로 생성된 fastq 파일에서 각각의 염기가 가지는 품질에 대한 수치를 의미합니다.
DNA, RNA에 대한 염기를 시퀀싱 기계로 읽어 들일 때, 필수적으로 에러가 발생할 수 밖에 없는데, quality score는 에러를 확률적으로 표시해 줍니다.
출처 : https://bgreat.tistory.com/152
특정 염기가 90% 확률의 정확도를 가진다면, Phred score를 10을 가지며, 99%의 정확도라면 20의 값을 가집니다. 주로 시퀀싱의 품질 기준으로 활용되는 Phred score 30에 대한 값은 염기에 대한 99.9% 정확도를 의미합니다.
염기가 확률을 가진다는 의미는 chemical signal을 digital 신호로 바꾸는 과정에서 오차가 생길 수 있기 때문입니다. 예를 들어, cluster 단위에서는 만약, A가 붉은색, G가 노란색인 경우 cluster의 색이 아주 약간의 노란색이 섞인 붉은색이 관찰된다면 이를 100% A라고 할 수 없기 때문입니다.
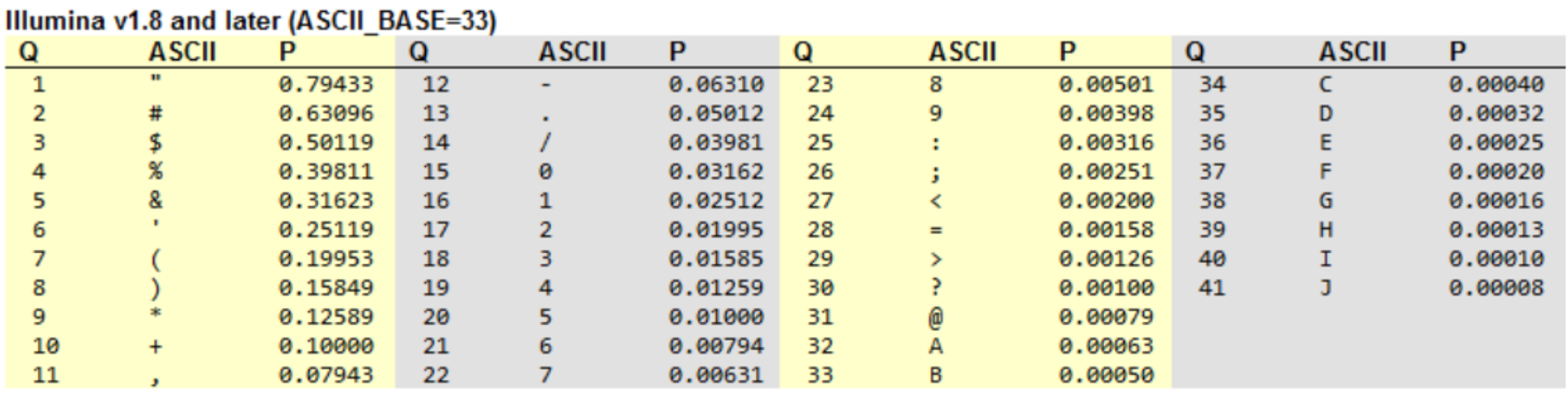
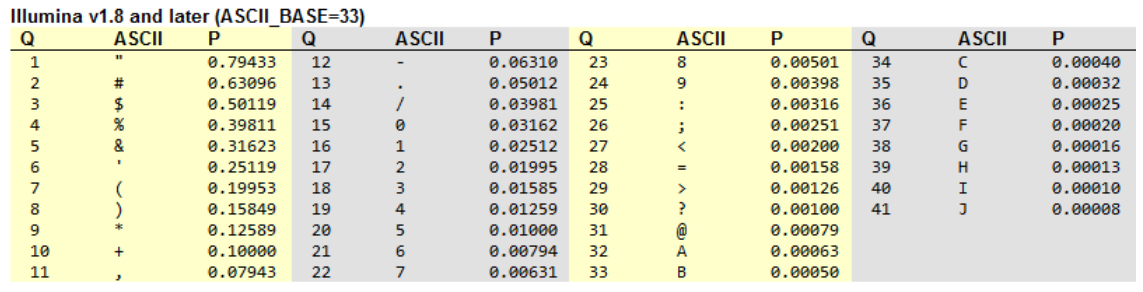
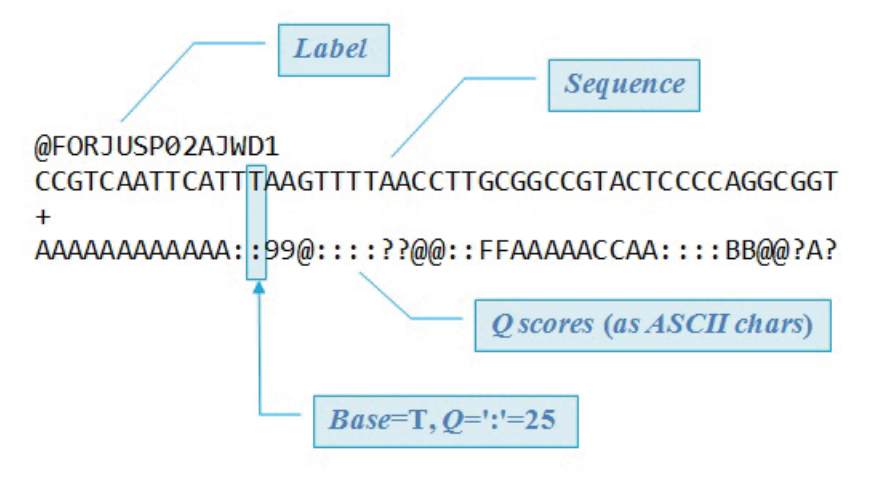
각 염기의 확률에 대한 Phred score 값은 숫자 2개로 표현이 가능하나, 각 염기가 한 자리(A,T,G,C)로 표현되기 때문에 Phred score 숫자를 ASCII CODE로 변환하여 한 자리로 표현합니다.
출처 : https://www.drive5.com/usearch/manual7/quality_score.html
여기서 Q 값은 Phred score 값을 의미하며, ASCII CODE는 Phred score 숫자를 한 자리로 표현한 값이며, P는 base에 대한 시퀀싱 오류가 발생할 확률입니다.
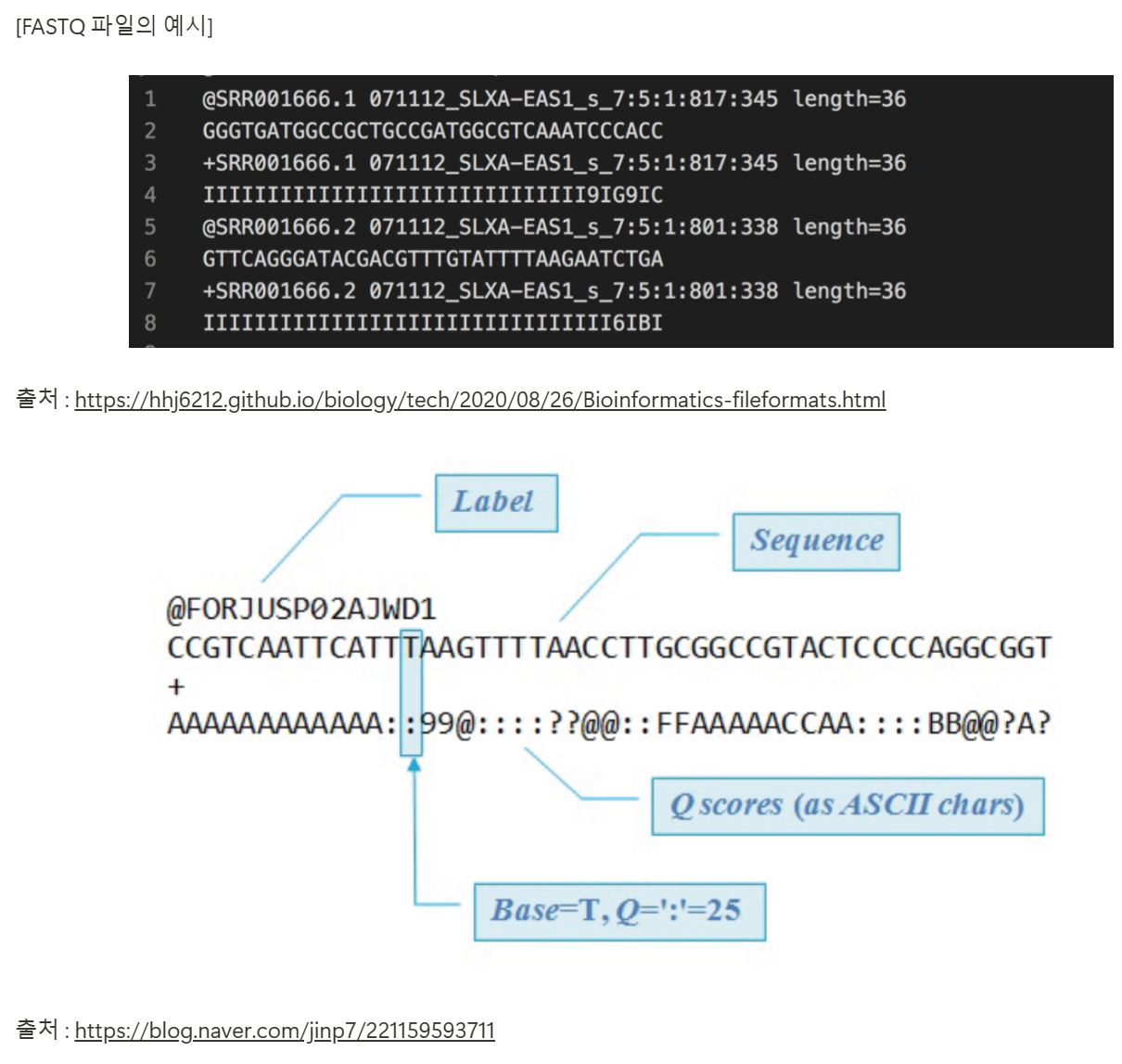
각 시퀀싱 리드(read)의 염기서열과 Phred 점수를 같이 표시한 것을 FASTQ 파일이라 부릅니다. FASTQ 파일은 시퀀스 고유ID, 각 리드의 시퀀스, “+” 기호, 시퀀스의 각 베이스 별 Phred 점수로 된 네 개의 줄로 구성되있습니다. Phred 점수는 ASCII 심볼로 표시합니다.
소개
FASTA 파일은 특정 분자의 서열을 나타내는 데 주로 사용됩니다.
주로 Genome 의 각 chromosome 에 대한 서열을 저장하는 데에 사용되거나, Gene / Transcript / Protein 각각의 염기 서열 및 아미노산 서열을 저장할 때 사용하는 형식입니다.
FASTA 파일의 형태는 NCBI FASTA format의 기준대로 다음과 같이 구성되어 있습니다.
먼저 첫 번째 줄은 “>”에 서열의 이름 / ID 가 기술되며, 주로 “헤더”라고 불립니다.
헤더 다음으로는 그 이름에 해당하는 서열이 나오게 됩니다.
DNA 서열 및 아미노산 모두 한 글자 문자열로 표현되며 일반적으로 한 줄에 서열의 길이가 80자를 넘지 않습니다.
FASTA 입력 중간에는 빈 줄이 허용되지 않습니다.
[FASTA 파일의 예시]

출처 : https://hhj6212.github.io/biology/tech/2020/08/26/Bioinformatics-fileformats.html
서열(Sequence) 의 표현
서열은 DNA 서열 및 단백질 서열일 수 있으며, 갭(gap) 또는 정렬 문자를 포함할 수 있습니다.
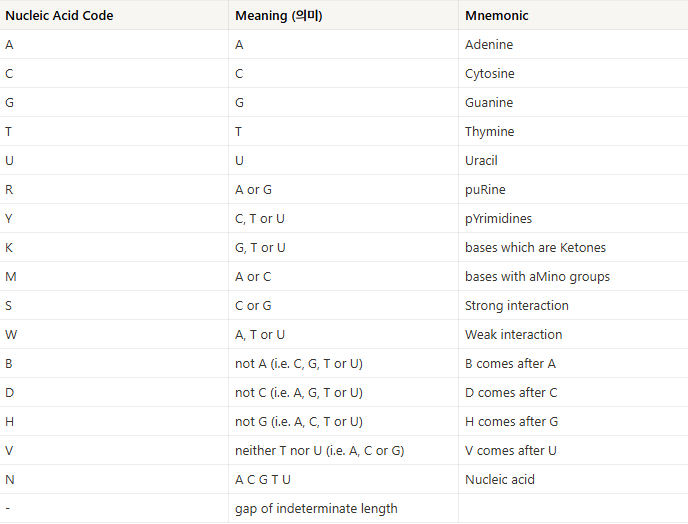
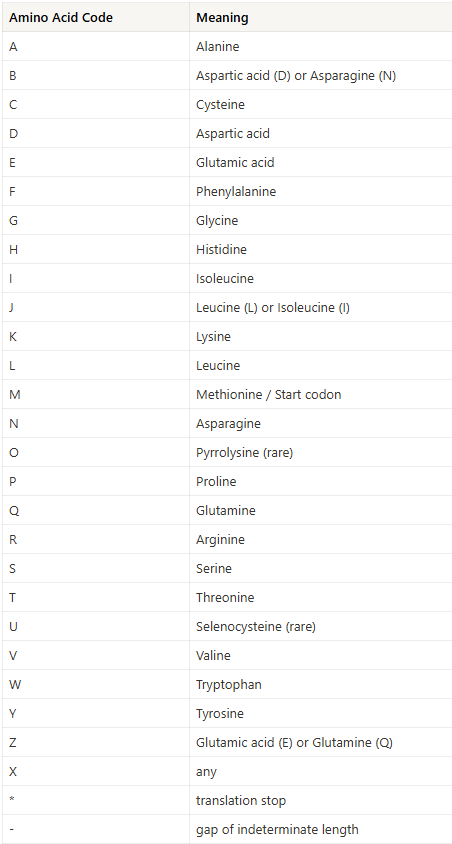
서열은 주로 표준 IUB/IUPAC 의 DNA 및 PROTEIN 코드로 표현됩니다.
[Nucleic Acid 테이블]
[Amino acid 테이블]
FASTA 파일의 확장자
FASTA 형식의 서열 데이터를 포함하는 텍스트 파일에 대한 표준 파일 확장자는 없습니다.
하지만, 일반적으로 아래 표와 같이 확장자와 그 의미를 사용합니다.

소개
FASTQ 파일은 생물학적 서열 데이터와 그 서열의 각 Base 별 품질 점수를 모두 저장하기 위한 텍스트 기반 형식입니다.
주로 NGS (Next generation sequencing data)의 결과를 저장하는 데에 사용됩니다.
즉, NGS 실험을 통한 결과로 cDNA library 서열을 읽어서 데이터를 얻으며, 각 cDNA Library의 염기서열을 알 수 있는데, 그 하나하나의 서열을 “read”라고 표현합니다.
FASTQ 파일은 여러 개의 리드(read) 정보를 한 파일에 저장하게 됩니다.
FASTQ 파일의 형식
FASTQ 파일은 각 서열 당 4개의 줄로 구성되어 있습니다.
- 첫 번째 줄 (sequence ID) : 주로 @로 시작되며 해당 서열의 이름을 나타냅니다.
- 두 번째 줄 (sequence) : 실제로 읽은 염기서열 정보입니다.
- 세 번째 줄 (description) : 주로 “+” 글자 만을 입력하거나 sequence ID 혹은 설명을 포함합니다.
- 네 번째 줄 (quality) : 각 염기서열이 얼마나 정확히 읽어진 것 인지를 나타냄, 주로 Phred quality score 값으로 표현됩니다.
[FASTQ 파일의 예시]
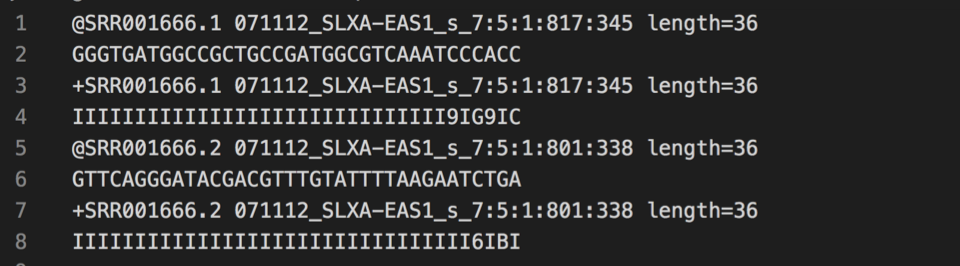
출처 : https://hhj6212.github.io/biology/tech/2020/08/26/Bioinformatics-fileformats.html
01-1. Quality Control
FastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/)
FastQC 프로그램은 NGS 결과 생성된 FASTQ 파일을 입력하여 데이터의 시퀀싱 품질을 평가하는 기능을 제공하는 소프트웨어로, 각 염기별 시퀀싱 품질, GC 컨텐츠, 어댑터 서열 등 원시 데이터에 대한 여러 품질 지표를 그래픽으로 시각화하여 제공합니다. 이를 통해, 사용자는 시퀀싱 데이터의 품질을 직관적으로 확인하고, 필요한 전처리 작업을 결정할 수 있습니다.
주요 활용되는 지표로 Per Base Sequence Quality, Per Sequence Quality Scores, Per Base Sequence Content, Per Base GC Content 등이 있습니다.
-
Input
- 각 샘플에 대한 raw data file (forward, reverse read)
- HG002_1.fastq.gz, HG002_2.fastq.gz
- HG003_1.fastq.gz, HG003_2.fastq.gz
- HG004_1.fastq.gz, HG004_2.fastq.gz
-
Practice
### FASTQC ### /root/bin/fastqc \ -t 8 \ --outdir ${RESULT_DIR} \ HG002_1.fastq.gz \ /root/bin/fastqc \ -t 8 \ --outdir ${RESULT_DIR} \ HG002_2.fastq.gz \ -
Option Description
-o / --outdir [DIR] fastQC 결과 파일을 생성할 디렉터리의 경로 입력 -t / --threads [NUM] 분석 수행을 위하여 사용하는 threads의 수 입력 -
Output
- 각 read 에 대한 fastqc 의 html 및 zip 파일 생성됨
- HG002_1_fastqc.html
- HG002_1_fastqc.zip
- HG002_2_fastqc.html
- HG002_2_fastqc.zip
02. Trimming
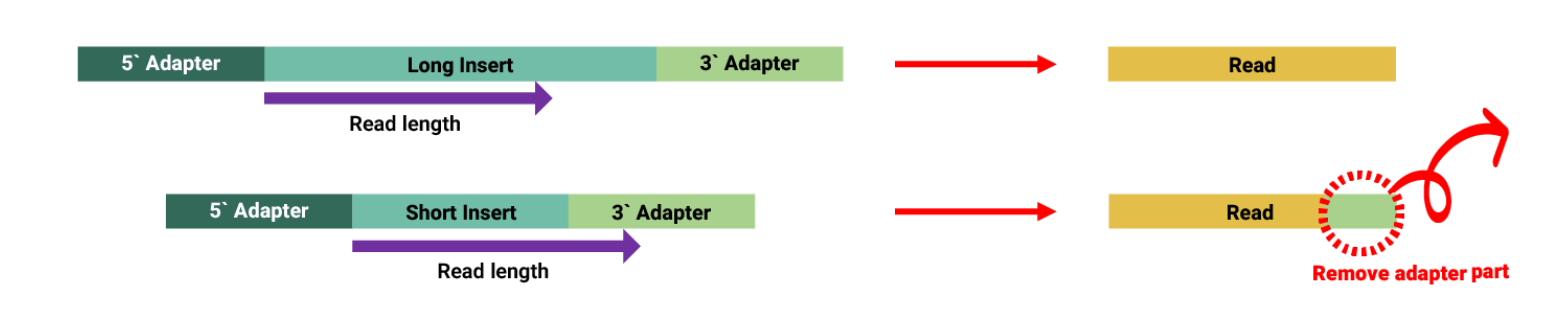
Trimming은 시퀀싱 데이터 분석의 품질을 높이기 위해 데이터를 정제하는 과정으로, 각 read 말단에서 나타나는 정확도가 낮은 부분을 잘라내고, read의 정확도 점수 평균이 너무 낮거나 염기 서열이 정확히 판정되지 않아 N으로 기록된 위치가 많은 경우 read 전체를 제거합니다.
또한, 라이브러리 제작 과정에서 내부 DNA 서열 길이가 짧은 경우 시퀀싱 장비가 내부 서열을 다 읽은 후 NGS 어댑터 서열을 읽게 되는데, 이 어댑터 부분은 원래 존재하지 않는 서열이므로 분석에 오류를 발생시킬 수 있습니다. 그러므로 read 말단부에서 어댑터(Adapter) 서열이 인식되는 경우 해당 서열을 제거합니다.
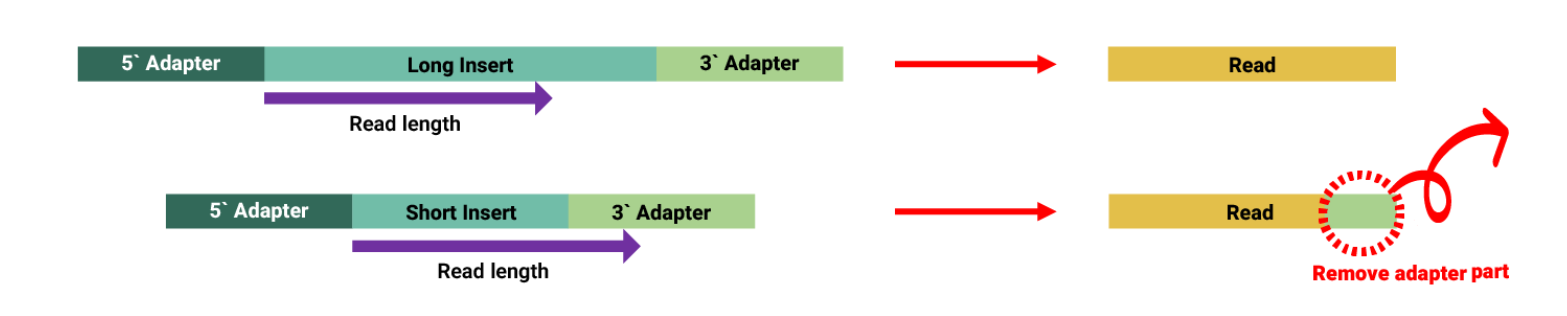
출처 : https://www.celemics.com/ko/resources/blogs/bioinformatics-standard-ngs-data-analysis-pipeline-2/
02-1. Trimming
Trimmomatic (https://github.com/usadellab/Trimmomatic)
원시 데이터에 대한 어댑터 시퀀스 제거 및 품질 낮은 서열을 판독하여 최소 품질 임계값(threshold)을 설정하고 이 수치 아래로 떨어지는 모든 염기를 제거하여 수행합니다.
python 기반인 cutadapt와 달리, Trimmomatic은 java 기반으로 개발되었으며 sickle과 동일하게 슬라이딩 윈도우 알고리즘을 사용하여 고품질의 서열만을 남기는 방식으로 수행됩니다.
-
Input
- 각 샘플에 대한 RAWDATA FILE (forward, reverse read)
- FORWARD_READ : HG002_1.fastq.gz
- REVERSE_READ : HG002_2.fastq.gz
-
Practice
java -jar \ ${TRIMMOMATIC_PATH}/trimmomatic-0.39.jar \ PE \ -threads 16 \ -phred33 \ ${RAW_DIR}/HG002_1.fastq.gz \ ${RAW_DIR}/HG002_2.fastq.gz \ ${RESULT_DIR}/HG002.R1.Trim.Paired.fastq.gz \ ${RESULT_DIR}/HG002.R1.Trim.UnPaired.fastq.gz \ ${RESULT_DIR}/HG002.R2.Trim.Paired.fastq.gz \ ${RESULT_DIR}/HG002.R2.Trim.UnPaired.fastq.gz \ ILLUMINACLIP:${ADAPTER_DIR}/TruSeq3-PE.fa:2:30:10 \ LEADING:3 TRAILING:3 SLIDINGWINDOW:4:15 MINLEN:36 -
Option description
[SE | PE] 데이터가 single end면 SE, paired end면 PE 선택 -phred33 Phred score 33 ILLUMINACLIP:[FILE]:[NUM]:[NUM]:[NUM] {Adapter 정보가 있는 파일}: {maximum mismatch counts}: {paired end에서 adapter reads간의 일치도}: {single end에서 adapter reads 간의 일치도} LEADING:[NUM] Read의 시작 부분에서 base를 제거할 최소 품질 기준 TRAILING:[NUM] Read의 끝 부분에서 base를 제거할 최소 품질 기준 SLIDING WINDOW:[NUM] {window size}:{평균 quality}, window를 이동시켜 평균 quality 이하로 내려가는 read 제거 MINLEN:[NUM] 유지되어야 할 최소 reads 길이 -
Output
- 각 샘플별 trimming 이후 결과 파일 (R1, R2에 대한 Paired, UnPaired read)
- HG002.R1.Trim.Paired.fastq.gz
- HG002.R1.Trim.UnPaired.fastq.gz
- HG002.R2.Trim.Paired.fastq.gz
- HG002.R2.Trim.UnPaired.fastq.gz
03. Alignment
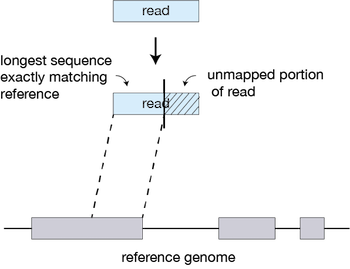
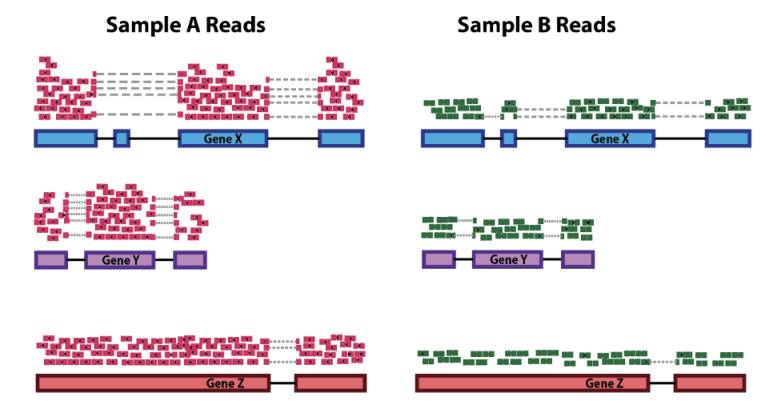
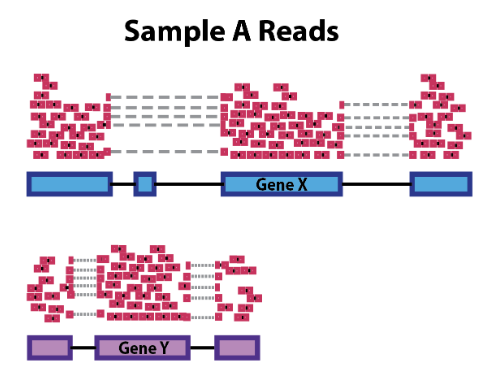
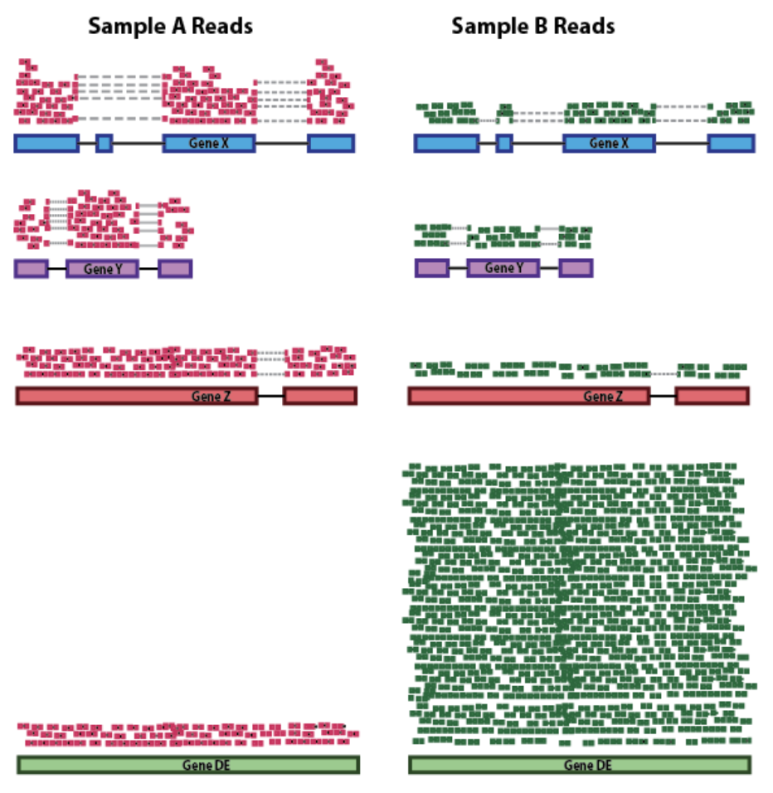
시퀀싱을 수행한 결과, 샘플별로 생성된 FASTQ 파일은 매우 많고 길이가 짧은 리드(read)로 구성되어 있습니다. 이러한 시퀀싱 리드들은 분석 대상 DNA 조각의 염기서열 정보를 나타내지만 어떤 염색체, 어느 위치에 있는 DNA 인지에 대한 정보는 담고 있지 않습니다. 따라서, 각 서열의 위치를 표준 유전체(reference genome)에서 찾아주는 작업이 필요하며, 이를 매핑(mapping) 혹은 정렬(alignment)이라고 합니다.
FASTQ 파일에 대한 매핑이 완료되면 각 시퀀싱 리드 별로 표준 유전체에서의 염색체 번호 및 위치가 기록됩니다. 이 정보를 담고 있는 파일을 SAM (sequence alignment map)이라고 하며, 일반적으로 용량이 크기 때문에 binary 형태로 압축된 BAM(binary alignment map) 파일을 많이 사용합니다.
SAM 혹은 BAM 파일에는 유전체 위치뿐만 아니라 매핑의 정확도를 나타내는 점수(mapping quality, MAPQ), 시퀀싱 리드에서 표준유전체 서열과 다른 염기를 표시해주는 정보(CIGAR string), paired-end 시퀀싱에서 같은 가닥의 반대편 시퀀싱 리드(mate)의 정보 등이 기록됩니다.
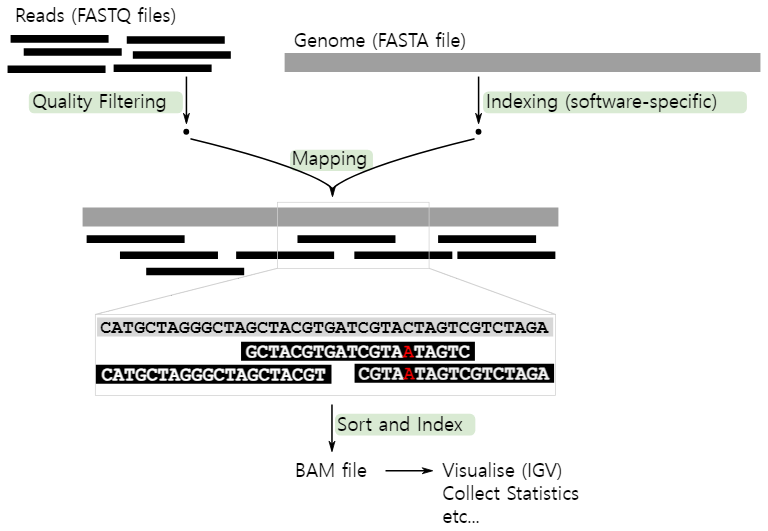
출처 : https://avantonder.github.io/Ghana_course/materials/01-intro_wgs/1.1-intro_wgs.html
-
정의
-
SAM 파일(Sequence alignment Map)이란 DNA 서열인 FASTQ 파일을 Reference genome 에 정렬했을 때 만들어지는 파일로
SAM 파일에서는 염기서열과 Reference에서의 위치 정보를 알 수 있습니다.
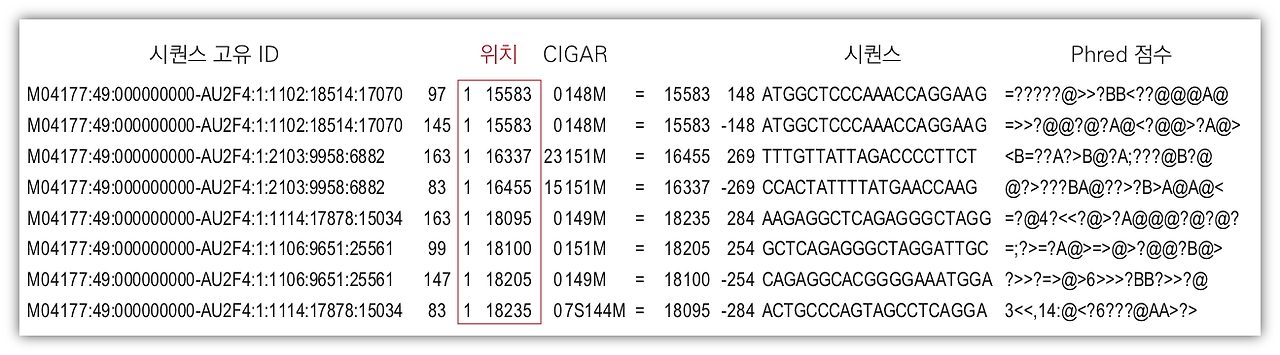
-
SAM 파일은 Header 부분과 Alignment 부분으로 이루어져 있습니다.
-
Header
- header 부분은 파일에 대한 설명이 포함됩니다 “@”로 시작되는 라인을 의미합니다
-
Alignment
- Alignment 는 각 read 에 대한 alignment 정보를 제공하는 부분입니다.
-
필수적인 11개의 column 을 구성되어 있으며 상황에 따라 몇 개의 column이 추가될 수 있습니다.
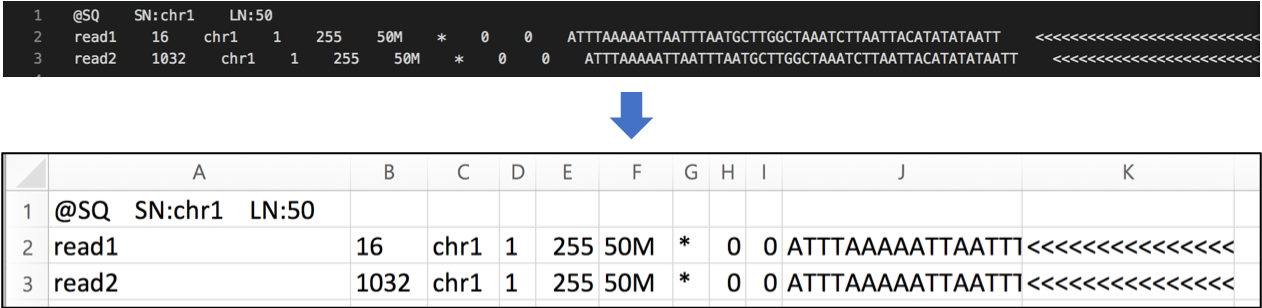
출처 : https://hhj6212.github.io/biology/tech/2021/06/13/samtools-flagstat.html

-
각 column에 대한 설명
1. QNAME : read 이름입니다.
2. FLAG: 2진수로 된 read alignment 에 대한 설명입니다.

3. RNAME: refernce sequence 의 이름입니다.
4. POS: reference sequence 에서 align 된 위치입니다.
5. MAPQ: mapping quality. 즉 얼마나 정확히 align 되었는지 수치료 표현합니다.
6. CIGAR string: alignment 정보를 표현한 문자열로 Match, Gap 등의 설명을 각 염기마다 표현합니다.

7. RNEXT: 다음 read 의 reference sequence 이름. 주로 paired end read 에 대한 분석을 위해 사용됩니다.
8. PNEXT: 다음 read 의 align 된 위치. 주로 paired end read 에 대한 분석을 위해 사용됩니다.
9. TLEN: Template length. paired-end read 둘의 left-end 부터 right-end 까지의 길이입니다.
10. SEQ: segment sequence. 염기 서열을 나타냅니다.
11. QUAL: Phread quality score 입니다.
- BAM(Binary Alignment Map) 파일은 SAM 파일과 동일한 정보를 가지고 있으며, 서로 변환이 가능합니다. 단, 정보를 압축하기 위해 Binary 형태로 저장하였다는 특징을 가지고 있습니다.
-
Header
-
SAM 파일(Sequence alignment Map)이란 DNA 서열인 FASTQ 파일을 Reference genome 에 정렬했을 때 만들어지는 파일로
SAM 파일에서는 염기서열과 Reference에서의 위치 정보를 알 수 있습니다.
03-1. Alignment (Mapping)
BWA (https://github.com/lh3/bwa)
Burrows-Wheeler Aligner(BWA) 알고리즘은 인간 유전체 매핑에 가장 많이 활용되는 알고리즘 중 하나로 BWA-ALN, BWA-SW 및 BWA-MEM 등의 방법으로 구분할 수 있습니다.
BWA-MEM은 70 bp 정도 되는 짧은 길이부터 1 Mb에 이르는 긴 길이의 시퀀싱 리드까지 빠르고 정확하게 매핑이 가능한 것으로 알려져 있습니다.
-
Input
- 샘플별 trimming 이후 결과 파일 (R1, R2에 대한 Paired read)
- HG002.R1.Trim.Paired.fastq
- HG002.R2.Trim.Paired.fastq
-
Practice
${BWA_PATH}/bwa mem \ -t $NCPU \ ${REF_GENOME} \ ${TRIMMED_DIR}/HG002.R1.Trim.Paired.fastq \ ${TRIMMED_DIR}/HG002.R2.Trim.Paired.fastq \ | ${SAMTOOLS_PATH}/samtools view -bS -> ${RESULT_DIR}/HG002.bam ${SAMTOOLS_PATH}/samtools sort \ -@ $NCPU \ -o ${RESULT_DIR}/HG002.sorted.bam \ ${RESULT_DIR}/HG002.bam ${SAMTOOLS_PATH}/samtools index \ -@ $NCPU \ ${RESULT_DIR}/HG002.sorted.bam -
Option description
-t nThreads 멀티스레드 처리를 위해 사용할 thread 수를 지정 -k minSeedLen seed 길이를 지정, 기본 값은 19 -R RGline 리드 그룹을 지정 -c maxOcc 최대 정렬할 리드 수를 지정 -B mmPenalty 갭 페널티(gap open penalty)을 설정, 기본 값은 4 -
Output
- HG002.sorted.bam : sorting 된 BAM 파일
- HG002.sorted.bai : sorting 된 BAM 파일의 인덱스
-
참고 알고리즘
참고 링크 : https://bioinformaticsandme.tistory.com/2
인간 유전체 분석에서 alignment 과정은 매우 복잡한 작업입니다.
시퀀싱을 통해 얻은 read(리드)로 약 30억 개의 전체 염기서열과 매칭하는 것은 쉽지 않습니다. 이로 인해 효율적이고 정확한 매칭을 수행하는 알고리즘과 도구가 필수적입니다.
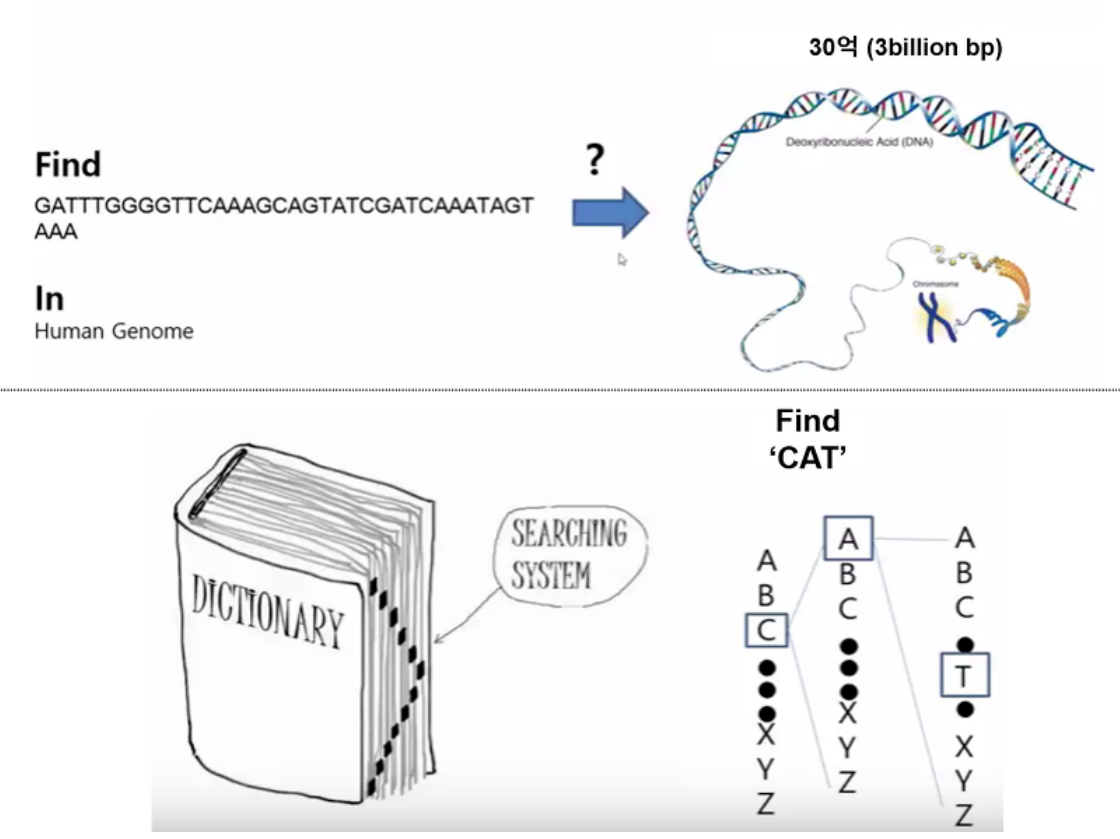
출처 : https://bioinformaticsandme.tistory.com/2
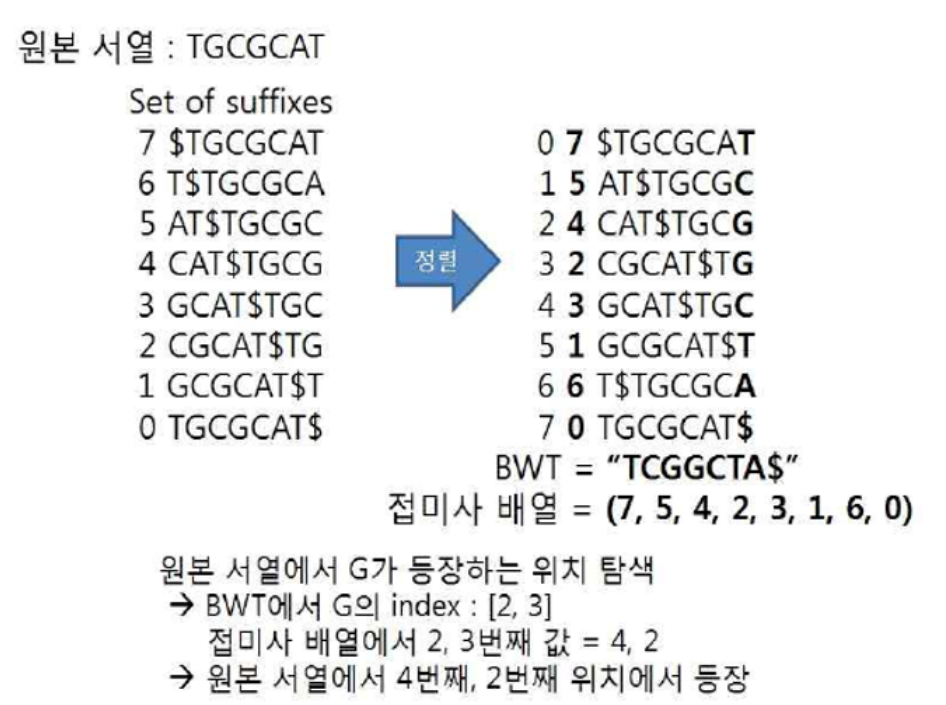
NGS 데이터를 alignment하는 프로그램으로 Bowtie와 BWA가 잘 알려져 있으며, 이들 프로그램은 Burrows-Wheeler Transform(BWT) 알고리즘을 주력으로 사용합니다.
-
BWT 알고리즘
- BWT 알고리즘은 문자열을 효율적으로 변환하는 방법입니다.
- 변환하고자 하는 문자열(BANANA)의 끝에 토큰($)을 추가합니다.
- 토큰($)을 포함한 문자열을 왼쪽으로 한 칸씩 이동하며 행렬을 생성합니다.
- 생성된 행렬의 각 행을 사전순으로 정렬(sorting)합니다(토큰은 맨 마지막 순서).
- 정렬된 행렬의 맨 마지막 열의 문자들로 변환된 문자열이 생성됩니다(BNN$AAA).
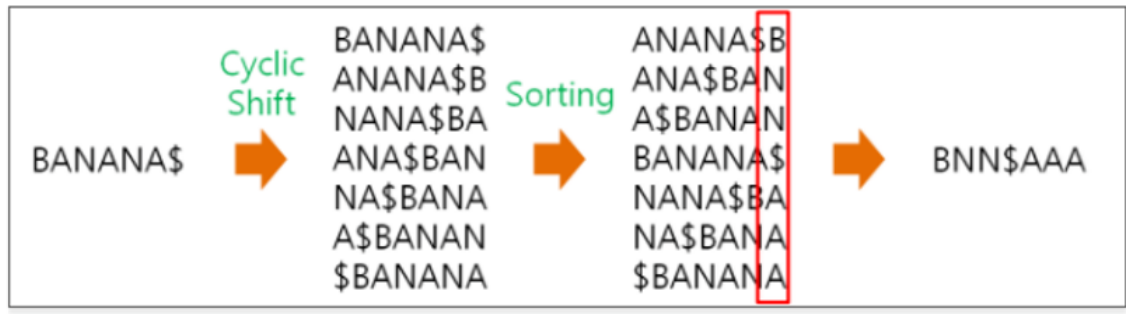
-
BWT 를 이용한 문자열 매칭
- BWT를 통해 원래 문자열을 복원하거나 특정 패턴을 검색할 수 있습니다.
- 원래 문자열은 토큰($)을 제외한 ROW 수와 동일하며, 글자 수를 확인할 수 있습니다.
- 첫 번째 컬럼의 문자는 마지막 컬럼의 바로 뒤에 위치한 문자입니다.
-
$ 토큰은 원래 문자열의 끝에 위치하므로 A가 마지막 글자임을 알 수 있습니다.
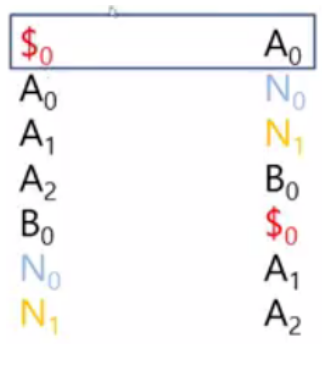
-
Last 컬럼과 First 컬럼의 문자 순서는 동일하며, 이를 통해 원래 문자열의 순서를 파악할 수 있습니다.
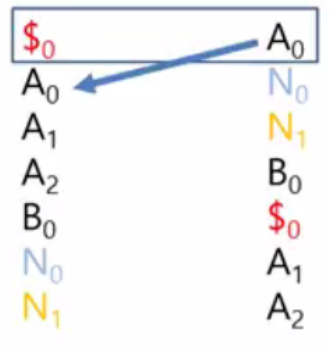
-
FM-Index
- Bowtie의 알고리즘인 FM-Index는 BWT 행렬의 Last-First Mapping 법칙을 이용하여 문자열 인덱스를 생성하는 방법입니다.
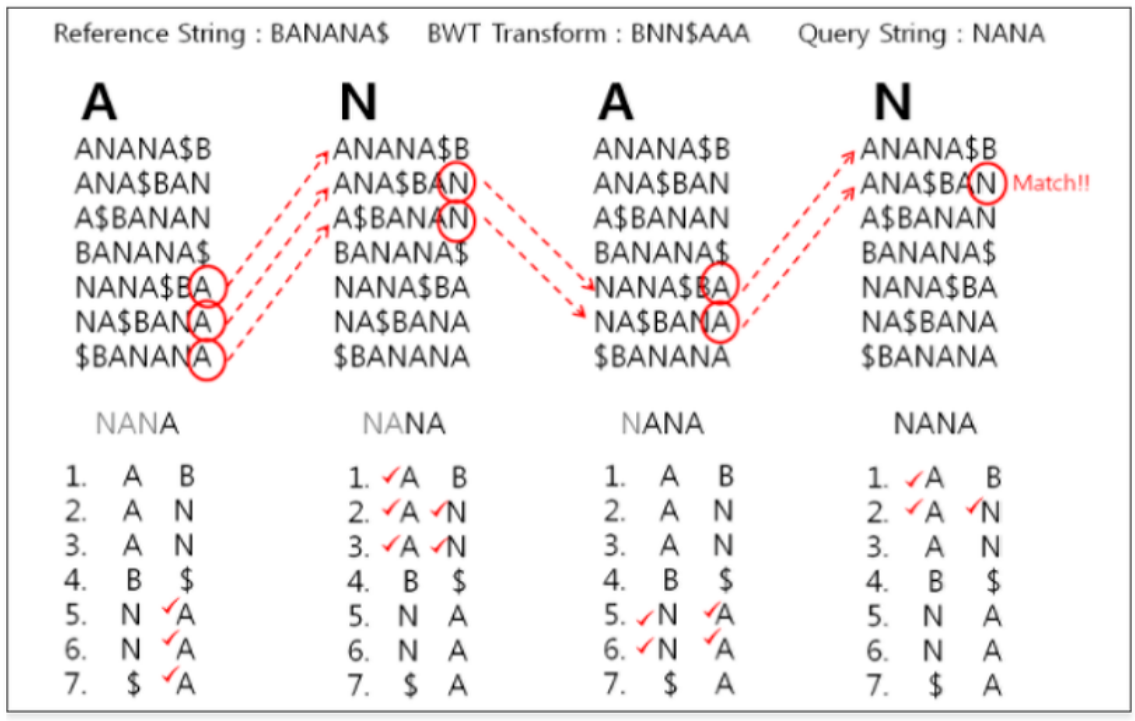
-
BWA (BWT + Suffix array)
-
BWA는 BWT와 접미사 배열(Suffix array)를 이용하여 정렬을 수행하는 알고리즘입니다.
-
BWA는 BWT와 접미사 배열(Suffix array)를 이용하여 정렬을 수행하는 알고리즘입니다.
-
BWT 알고리즘
04. Processed BAM file
앞에서 생성한 BAM 파일을 사용하여 샘플별로 참조 유전체 서열과 다른 정확한 변이(SNP, INDEL)을 발굴하기 위해서는 크게 3가지 단계의 절차가 필요합니다.
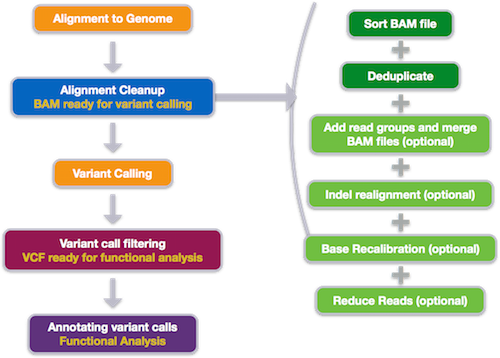
출처 : https://hbctraining.github.io/In-depth-NGS-Data-Analysis-Course/sessionVI/lessons/01_alignment.html
04-1. AddOrReplaceReadGroup
BAM 파일에 read group을 추가하고 coordinate 정렬을 수행합니다. 이를 통해 BAM 파일에 포함된 리드가 어떤 샘플에서 유래했는지 식별할 수 있게 해주며, 이후 분석 단계에서 필요한 read group의 정보를 BAM 파일에 포함시킵니다.
참고 링크 : https://gatk.broadinstitute.org/hc/en-us/articles/360037226472-AddOrReplaceReadGroups-Picard
-
Input
- HG002.sorted.bam : sorting 된 BAM 파일
- HG002.sorted.bai : sorting 된 BAM 파일의 인덱스
-
Practice
java -jar -Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU ${PICARD_PATH}/picard.jar \ AddOrReplaceReadGroups \ I=${BAM_DIR}/HG002.sorted.bam \ O=${RESULT_DIR}/HG002.sorted.RG.bam \ RGID=HG002 \ RGLB=lib.HG002 \ RGPL=illumina \ RGPU=HG002 \ RGSM=HG002 ${SAMTOOLS_PATH}/samtools index \ ${RESULT_DIR}/${sample_name}.sorted.RG.bam -
Option description
I / INPUT 입력할 BAM 또는 SAM 파일을 지정 O / OUTPUT 수정된 리드 그룹 정보를 포함한 출력 BAM 또는 SAM 파일을 지정 RGID 리드 그룹의 고유 ID를 지정 RGLB 시퀀싱에 사용된 라이브러리의 이름을 지정 RGPL 시퀀싱이 수행된 플랫폼을 지정 RGPU 시퀀싱 플랫폼 유닛을 지정 RGSM 시퀀싱된 샘플의 이름을 지정 -
Output
- HG002.sorted.RG.bam : Read group을 추가한 sorting 된 BAM 파일
- HG002.sorted.RG.bai : Read group을 추가한 sorting 된 BAM 파일의 인덱스
04-2. MarkDuplicates
PCR duplicate는 동일한 DNA 분자로부터 증폭된 산물들이 중복되어 포함된 것을 의미합니다. NGS 데이터에서 리드(reads)의 비율이 원래의 DNA 분자들과 최대한 비슷하게 유지되려면, PCR duplicate를 제거해주는 과정이 매우 중요합니다. 이는 duplication을 제거함으로써 변이 비율이 실제 DNA 분자 내에서의 변이 비율과 유사해 질 수 있습니다.
GATK의 MarkDuplicates 도구는 paired-end read의 위치 정보를 바탕으로 원본 DNA의 구조를 유추하여 중복된 데이터를 탐지하고 표시하는 역할을 합니다. 이 과정에서 read와 그 pair에 해당하는 두 개의 5’ position을 비교하여 중복 여부를 결정하며, BARCODE_TAG 옵션을 사용해 분자 바코드를 적용하면 중복 표시가 더 정확하게 이루어집니다. 중복된 리드를 식별한 후에는 각 리드의 base-quality score 값을 합산하여 primary read와 duplicate된 read를 구분합니다. 이를 통해 중복 리드를 정확하게 처리하여 분석의 신뢰성을 높일 수 있습니다.
참고 링크 : https://gatk.broadinstitute.org/hc/en-us/articles/360037052812-MarkDuplicates-Picard
-
Input
- HG002.sorted.RG.bam : Read group을 추가한 sorting 된 bam 파일
- HG002.sorted.RG.bai : Read group을 추가한 sorting 된 bam 파일의 인덱스
-
Practice
java -jar -Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU ${PICARD_PATH}/picard.jar \ MarkDuplicates \ I=${RESULT_DIR}/HG002.sorted.RG.bam \ O=${RESULT_DIR}/HG002.sorted.RG.dedup.bam \ M=${RESULT_DIR}/HG002.marked_dup_metrics.txt ${SAMTOOLS_PATH}/samtools index \ {RESULT_DIR}/HG002.sorted.RG.dedup.bam -
Option description
I / INPUT 중복 리드를 찾을 BAM 또는 SAM 파일을 지정 O / OUTPUT 중복 마킹이 완료된 후의 결과 BAM 파일을 지정 M / METRICS_FILE 중복 리드에 대한 통계 정보를 기록할 파일을 지정 REMOVE_DUPLICATES 중복으로 마킹된 리드를 파일에서 실제로 제거할지 여부를 설정, 기본값은 false CREATE_INDEX BAM 파일의 색인 생성 여부를 지정, 기본값은 false -
Output
- HG002.sorted.RG.dedup.bam : Read group을 추가하였으며 duplication이 제거된 sorting 된 bam 파일
- HG002.sorted.RG.dedup.bai : Read group을 추가하였으며 duplication이 제거된 sorting 된 bam 파일의 인덱스
04-3. BQSR (Base Quality Score Recalibration)
즉, 데이터에서 보고된 서열의 정확도 점수 분포를 분석하여 실험적으로 더 정확한 분포를 갖도록 재조정하는 단계로, 사용자가 입력한 Known SNP 위치 정보를 가지고 데이터의 에러율을 다시 계산(BaseRecalibrator)한 후, 이를 반영하여 정확도 점수를 재조정(ApplyBQSR)하는 두 단계로 수행됩니다.
참고 링크 : https://gatk.broadinstitute.org/hc/en-us/articles/360035890531-Base-Quality-Score-Recalibration-BQSR
-
Input
- HG002.sorted.RG.dedup.bam : Read group을 추가하였으며 duplication이 제거된 sorting 된 bam 파일
- HG002.sorted.RG.dedup.bai : Read group을 추가하였으며 duplication이 제거된 sorting 된 bam 파일의 인덱스
-
Practice
${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ BaseRecalibrator \ -I ${RESULT_DIR}/HG002.sorted.RG.dedup.bam \ -R ${REF_GENOME} \ --known-sites ${KNOWN_VARIANT_PATH}/dbsnp_138.hg38.vcf \ -O ${RESULT_DIR}/HG002.sorted.RG.dedup.recal_table ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ ApplyBQSR \ -R ${REF_GENOME} \ -I ${RESULT_DIR}/HG002.sorted.RG.dedup.bam \ --bqsr-recal-file ${RESULT_DIR}/HG002.sorted.RG.dedup.recal_table \ -O ${RESULT_DIR}/HG002.sorted.RG.dedup.BQSR.bam ${SAMTOOLS_PATH}/samtools index \ {RESULT_DIR}/HG002.sorted.RG.dedup.BQSR.bam -
Option description
-I 입력할 BAM 또는 CRAM 파일을 지정 -R 분석에 사용할 참조 유전체 파일을 지정 --known-sites 시퀀싱 오류를 고려하지 않고 실제 변이를 포함하는 사이트의 VCF 파일을 지정 -O 보정 후 생성될 recalibration 테이블 파일을 지정 -
Output
- HG002.sorted.RG.dedup.BQSR.bam : Read group을 추가하였으며 duplication이 제거되었고, BSQR을 통해 Base의 quality가 보정된 sorting 된 bam 파일
- HG002.sorted.RG.dedup.BQSR.bai : Read group을 추가하였으며 duplication이 제거되었고, BSQR을 통해 Base의 quality가 보정된 sorting 된 bam 파일의 인덱스
-
참고 알고리즘
참고 링크 : https://bioinformaticsandme.tistory.com/77
BQSR을 수행하기 위해 필요한 것은 잘 알려진 (Known) SNP VCF 파일입니다.
그 이유는 BQSR 알고리즘에서 dbSNP에 매칭되지 않는 base는 “에러”로 가정하기 때문입니다.
1) Finding ERROR
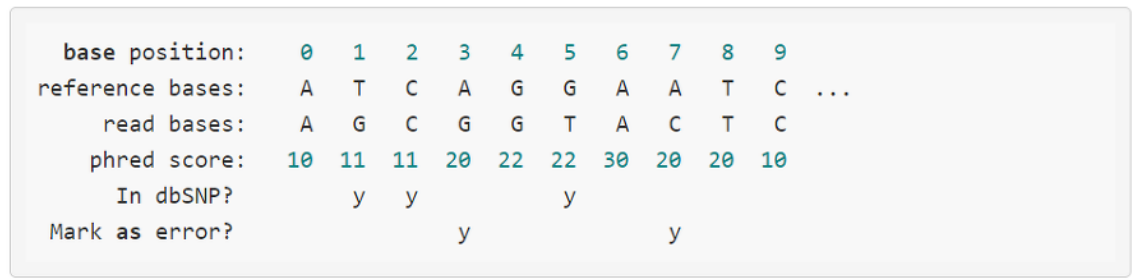
출처 : https://bioinformaticsandme.tistory.com/77
예제 처럼, 어떤 염색체 위에 0~9 BASE 까지 길이가 10인 리드가 존재한다고 가정합니다.
⇒ BQSR 에서 ERROR로 여겨지는 포지션은 3번과 7번입니다.
- 이는 3번과 7번이 read bases와 ref 에 대한 염기가 다르고, 알려진 db에도 존재하지 않기 때문입니다.
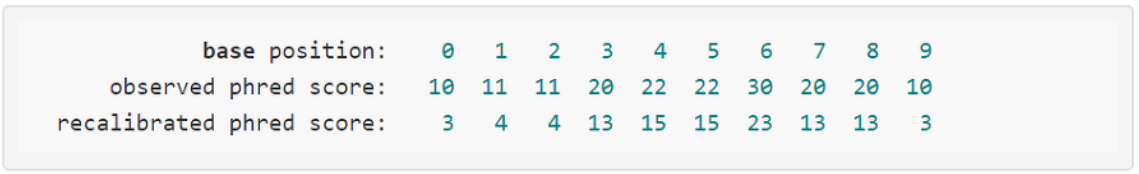
2) Aggregating the reported phred score
- 각 base 에 대한 quality score 값인 phred score 를 확률로 변환시킵니다. (0.1+0.079 + 0.079 + 0.01 + 0.006 + 0.006 + 0.001 + 0.01 + 0.01 + 0.01)/10 = 0.0401
- 이것을 다시 phred score 값으로 변환하면, phred score = -10*log10(0.0401) ~= 14 입니다..
- 따라서, 예제에 확인되는 read 에 대한 phred score는 약 14입니다.
3) Calculating the empirical phred score
- 10개의 염기 중에서 2개를 에러로 가정하였으므로, 시퀀스는 약 2/10 = 0.2의 에러 확률을 가집니다.
- 이를 phred score 값으로 변환하면 -10 * log10(0.2) ~= 7 (empirical phred score)
- empirical phred score 값이 7 정도의 에러가 있다면, 원래 관찰된 phred score 는 이미 +7 정도의 에러가 더해진 값이라고 생각 할 수 있으므로, phred score 값을 -7로 진행하여 recalibration 을 진행합니다.
05. Variant Calling
SAM/BAM 파일이 생성되면 각 시퀀싱 리드를 분석하여 특정 위치에서 표준 유전체 서열과 다른 변이(variation)가 있는지 찾아내는 변이 검출(variant calling) 작업을 수행합니다. 이 과정에서 한 위치에 여러 개의 시퀀싱 리드를 종합하여 변이가 있는지 확률적으로 판단하게 됩니다.
-
정의
-
VCF(Variant Call Format)는 유전적 변이를 저장하고 전송하기 위한 표준 파일 형식으로, 주로 DNA 시퀀스 데이터를 기반으로 한 변이 호출 정보를 담고 있습니다.
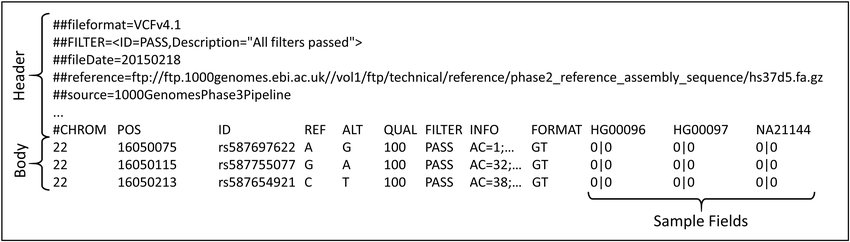
출처 : https://www.researchgate.net/figure/Sample-VCF-File-Structure_fig2_347069472
-
VCF 파일은 주석(header) 부분과 데이터 부분으로 나뉘어 있습니다.
- Header : 파일의 메타데이터와 형식 정보를 포함하며, 각 변이에 대한 정보를 정의하는 다양한 필드와 그 의미를 설명합니다.
- Body : 각 변이의 정보(예: 위치, 변이 유형 등)를 행 단위로 저장합니다.
-
각 변이에 대한 정보는 다음과 같은 필드를 포함합니다.

출처 : https://davetang.github.io/learning_vcf_file/
- CHROM: 변이가 발생한 염색체의 이름
- POS: 변이가 발생한 위치
- ID: 변이에 대한 고유 식별자
- REF: 참조 염기 서열
- ALT: 대체 염기 서열 (변이)
- QUAL: 변이에 대한 신뢰도 점수
- FILTER: 필터링 정보
- INFO: 추가적인 정보(예: 변이의 생물학적 영향 등)
- FORMAT: 샘플 데이터의 형식 정보를 제공하며, 각 샘플의 변이 정보를 포함.
-
VCF(Variant Call Format)는 유전적 변이를 저장하고 전송하기 위한 표준 파일 형식으로, 주로 DNA 시퀀스 데이터를 기반으로 한 변이 호출 정보를 담고 있습니다.
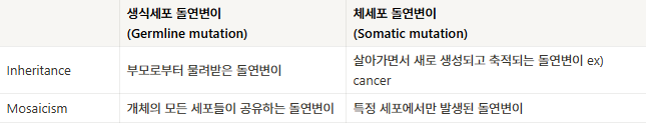
NGS 데이터를 이용하여 발굴할 수 있는 변이는 크게 두 가지로 구분할 수 있습니다.
바로 Germline mutation(생식세포 돌연변이)와 Somatic mutation(체세포 돌연변이)입니다.
05-1. Germline short variant (SNP, INDEL) 을 발굴하기 위한 단계
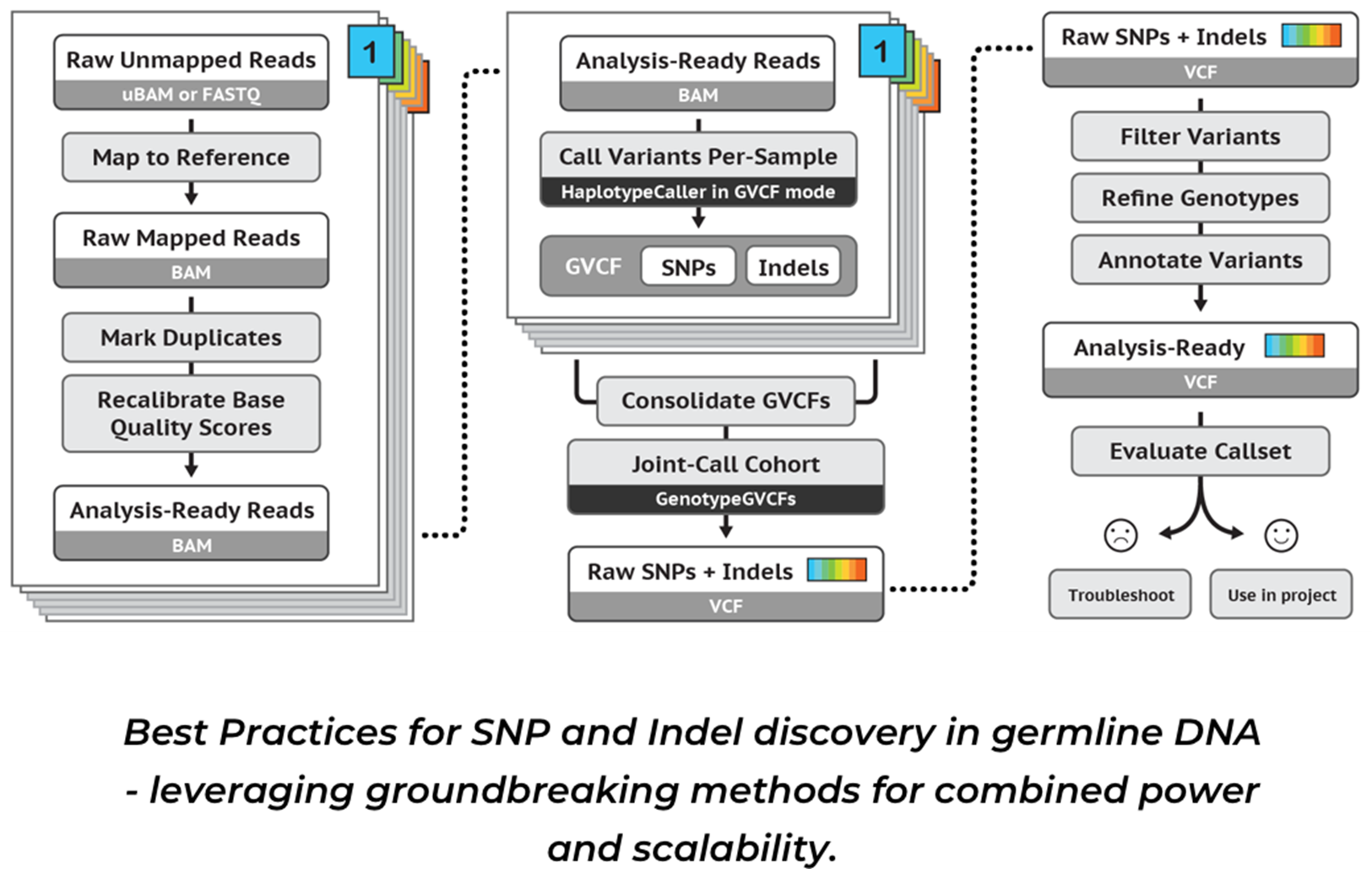
출처 : https://2wordspm.wordpress.com/2019/03/08/ngs-분석-파이프-라인의-이해-gatk-best-practice/
1) HaplotypeCaller (GVCF mode) : 변이 호출
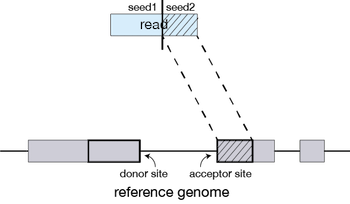
HaplotyperCaller 는 활성화된 영역에서 haplotype의 local denovo 어셈블리를 통해 SNP와 INDEL을 동시에 호출할 수 있습니다. 즉, 해당 프로그램이 변이에 대한 징후를 보이는 영역을 발견할 때마다 기존 매핑 정보를 폐기하고, 해당 영역에 매핑되는 리드(read)를 재조립하여 서로 가까이 있는 다른 유형의 변이에 대해 더 정확하게 호출할 수 있다는 장점을 가지고 있습니다.
GVCF mode 는 genetic VCF 형태로 no-variant block record를 포함하여 모든 position 에 대한 정보를 가지고 있습니다. 이를 통해 추가적인 분석을 통해 생성되는 샘플을 확장하고자 할 때 기존 VCF mode 에서는 N+1개 샘플에 대한 변이 호출을 모두 다시 수행해야 했다면 GVCF mode는 추가된 1개의 샘플만 분석을 진행하고 결과를 합치는 방향으로 분석을 수행할 수 있습니다.
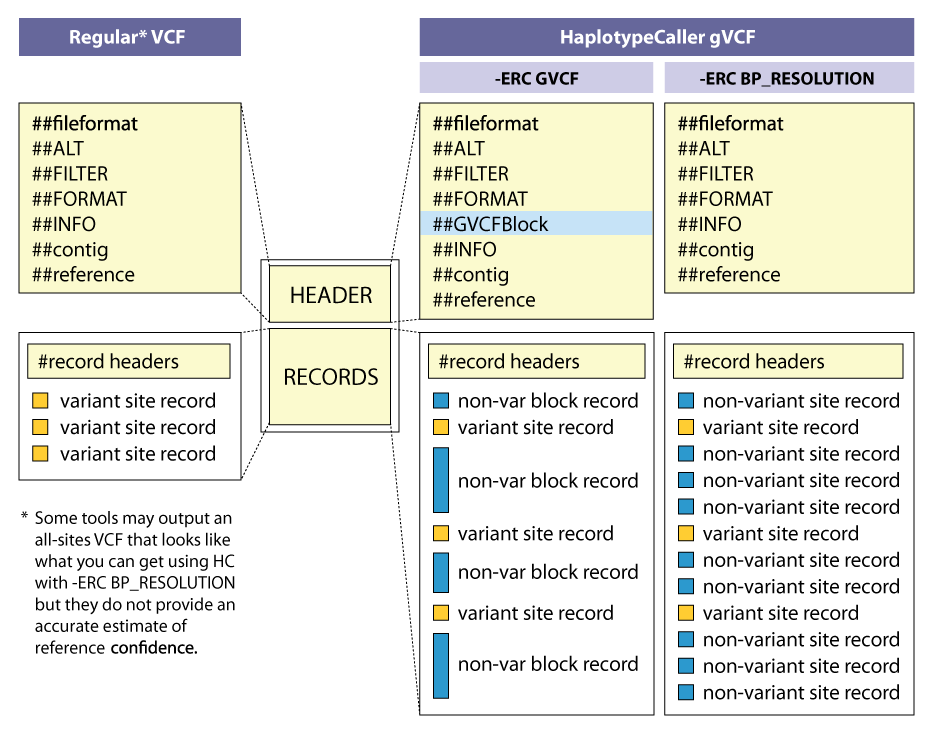
-
Input
- HG002.sorted.RG.dedup.BQSR.bam : Read group을 추가하였으며 duplication이 제거되었고, BSQR을 통해 Base의 quality가 보정된 sorting 된 bam 파일
- HG002.sorted.RG.dedup.BQSR.bai : Read group을 추가하였으며 duplication이 제거되었고, BSQR을 통해 Base의 quality가 보정된 sorting 된 bam 파일의 인덱스
-
Practice
${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ HaplotypeCaller \ -R ${REF_GENOME} \ -I ${BAM_DIR}/HG002.sorted.RG.dedup.BQSR.bam \ --emit-ref-confidence GVCF -O ${RESULT_DIR}/HG002.raw.g.vcf -
Option description
-I 변이를 호출할 BAM 또는 CRAM 파일을 지정 -R 분석에 사용할 참조 유전체 파일을 지정 --emit-ref-confidence 참조 서열에 대한 신뢰도 정보를 출력할 방법 설정 NONE 신뢰도 정보를 출력하지 않음 GVCF 신뢰도 정보를 포함하여 gVCF 파일로 출력 BP_RESOLUTION 각 염기 위치에 대한 신뢰도 정보를 출력 -O 호출된 변이 정보를 저장할 VCF 파일을 지정 -
Output
- 각 샘플별 GVCF 형태의 VCF 파일 : HG002.raw.g.vcf
2) GenotypeGVCFs : 샘플이 가진 변이 호출
HaplotyperCaller GVCF 모드를 통해 생성한 GVCF 파일은 일반 VCF 파일과는 달리, 각 샘플에서 변이 여부에 관계없이 모든 위치에 대한 정보를 포함하고 있으므로 특정 샘플이 가진 변이만을 호출하기 위해, GenotypeGVCFs를 수행해야 합니다.
-
Input
- 각 샘플별 GVCF 형태의 VCF 파일 : HG002.raw.g.vcf
-
Practice
${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ GenotypeGVCFs \ -R ${REF_GENOME} \ -V ${RESULT_DIR}/HG002.raw.g.vcf \ -O ${RESULT_DIR}/HG002.genotyped.vcf -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 -V 입력 GVCF 파일 (하나 이상의 GVCF파일을 지정할 수 있음) -O 출력할 VCF 파일을 지정 -
Output
- 특정 샘플이 가진 변이만을 포함한 VCF 형태의 파일 : HG002.genotyped.vcf
3) SNP, INDEL 파일로의 구분
사용자는 VCF 파일에서 SNP와 INDEL을 구분하거나 특정한 변이 타입만을 선택 및 필터링을 진행할 수 있습니다.
-
Input
- 특정 샘플이 가진 변이만을 포함한 VCF 형태의 파일 : HG002.genotyped.vcf
-
Practice
### Seperate SNP ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ SelectVariants \ -R ${REF_GENOME} \ -V ${RESULT_DIR}/HG002.genotyped.vcf \ --select-type-to-include SNP \ -O ${RESULT_DIR}/HG002.snp.vcf ### Seperate indel ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ SelectVariants \ -R ${REF_GENOME} \ -V ${RESULT_DIR}/HG002.genotyped.vcf \ --select-type-to-include INDEL \ -O ${RESULT_DIR}/HG002.indel.vcf -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 -V 입력 GVCF 파일 --select-type-to-include VCF 파일에서 선택하고자 하는 변이 (예: SNP, INDEL) -O 출력할 VCF 파일을 지정 -
Output
- 특정 샘플이 가진 SNP, INDEL 변이를 포함하는 VCF 파일 : HG002.snp.vcf, HG002.indel.vcf
4) Hard filtering
GATK Hard Filtering은 변이 호출 후 품질 기준에 따라 변이를 필터링하는 방법입니다. 사용자가 지정한 값에 따라 변이의 품질을 평가하고 품질이 낮은 변이를 "filtered"로 표시하여 결과에서 제외할 수 있게 해줍니다.
-
Input
- 특정 샘플이 가진 SNP, INDEL 변이를 포함하는 VCF 파일 : HG002.snp.vcf, HG002.indel.vcf
-
Practice
### Apply the filter to SNP ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ VariantFiltration \ -R ${REF_GENOME} \ -V ${RESULT_DIR}/HG002.snp.vcf \ --filter-expression "QD < 2.0 || FS > 60.0 || MQ < 40.0 || MQRankSum < -12.5 || ReadPosRankSum < -8.0" \ --filter-name "HARD_FILTER" \ -O ${RESULT_DIR}/HG002.snp.hard_filtered.vcf ### Apply the filter to indel ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ VariantFiltration \ -R ${REF_GENOME} \ -V ${RESULT_DIR}/HG002.indel.vcf \ --filter-expression "QD < 2.0 || FS > 200.0 || ReadPosRankSum < -20.0" \ --filter-name "HARD_FILTER" \ -O ${RESULT_DIR}/HG002.indel.hard_filtered.vcf -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 -V 입력 GVCF 파일 --filter-name 필터 기준을 충족하지 못하는 변이에 적용할 이름 --filter-expression 필터링 기준을 정의하는 식(expression). -O 필터링된 결과에 대한 VCF 파일을 지정 -
Output
- 사용자의 기준에 따라 필터링이 완료된 특정 샘플이 가진 SNP, INDEL 변이를 포함하는 VCF 파일 : HG002.snp.hard_filtered.vcf, HG002.indel.hard_filtered.vcf
05-2. Somatic short variant (SNP, INDEL) 을 발굴하기 위한 단계
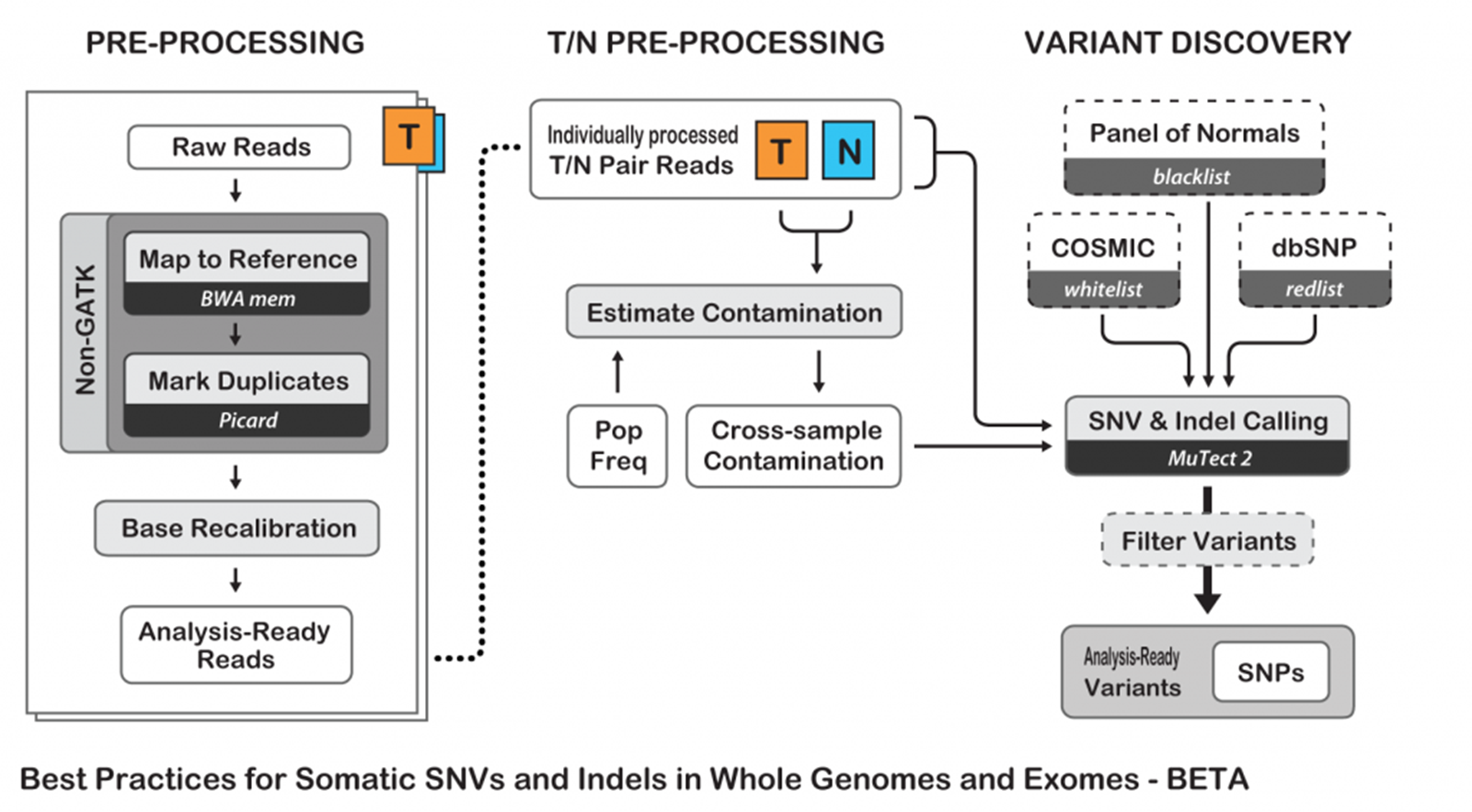
0) CreateSomaticPanelOfNormals
Somatic variant calling 분석 시작 전, germline 및 artifact variant를 제거하기 위해 Panel Of Normal (PON)을 생성하는 과정으로, 하나 이상의 Normal sample을 대상으로 Mutect의 tumor-only mode를 수행하여 생성합니다.
-
Input
- ${CONTROL_1}.sorted.RG.dedup.BQSR.bam : normal_1 샘플에 대한 bam 파일
- ${CONTROL_2}.sorted.RG.dedup.BQSR.bam : normal_2 샘플에 대한 bam 파일
-
Practice
### PON File 생성 ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ Mutect2 \ -R ${REF_GENOME} \ -I ${NORMAL_BAM_DIR}/${CONTROL_1}.sorted.RG.dedup.BQSR.bam \ -tumor ${CONTROL_1} \ --disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \ --germline-resource ${DB_PATH}/af-only-gnomad.raw.sites.hg19.vcf.gz \ -O ${CONTROL_1}.PON.vcf.gz ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp \ Mutect2 \ -R ${REF_GENOME} \ -I ${NORMAL_BAM_DIR}/${CONTROL_2}.sorted.RG.dedup.BQSR.bam \ -tumor ${CONTROL_2} \ --disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \ --germline-resource ${DB_PATH}/af-only-gnomad.raw.sites.hg19.vcf.gz \ -O ${CONTROL_2}.PON.vcf.gz ### CreateSomaticPanelOfNormals ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ CreateSomaticPanelOfNormals \ --vcfs ${CONTROL_1}.PON.vcf.gz \ --vcfs ${CONTROL_2}.PON.vcf.gz \ -O ${NORMAL_BAM_DIR}/${CONTROL}.PON.vcf.gz -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 -I 분석할 BAM 또는 CRAM 파일을 지정 -tumor 분석할 종양 샘플의 ID를 지정 --disable-read-filter 기본적으로 활성화된 리드 필터 중 일부를 비활성화 --germline-resource 체세포 변이를 탐지할 때 참조할 알려진 생식세포 변이(germline variant) 리소스 파일을 지정 -O 필터링된 변이 결과를 저장할 VCF 파일을 지정 -
Output
- ${CONTROL}.pon.vcf.gz : Normal sample에 대한 Panel Of Normal (PON) 파일
1) Variant calling (Mutect2)
HaplotypeCaller와 마찬가지로, Mutect2는 활성 영역에서 haplotype의 로컬 de-novo 어셈블리 방식을 통해 SNV와 indel을 동시에 호출합니다. 즉, Mutect2가 체세포 변이의 징후를 보이는 영역을 발견하면 기존 매핑 정보를 폐기하고 해당 영역에서 판독을 완전히 재조립하여 후보 변이에 대한 haplotype을 생성합니다. Mutect2는 Pair-HMM 알고리즘을 통해 각 판독을 각 haplotype에 맞춰 likelihood 행렬을 얻습니다. 마지막으로 Bayesian somatic likelihoods 모델을 적용하여 시퀀싱 오류에 비해 대립유전자가 체세포 변이가 될 log odds를 얻습니다.
-
Input
- ${CONTROL}.pon.vcf.gz : Normal sample에 대한 Panel Of Normal (PON) 파일
- ${sample_name}.sorted.RG.dedup.BQSR.bam : Read group을 추가하였으며 duplication이 제거되었고, BSQR을 통해 Base의 quality가 보정된 sorting 된 bam 파일
-
Practice
${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ Mutect2 \ -R ${REF_GENOME} \ -I ${TUMOR_BAM_DIR}/${sample_name}.sorted.RG.dedup.BQSR.bam \ -tumor ${sample_name} \ -I ${NORMAL_BAM_DIR}/${CONTROL}.sorted.RG.dedup.BQSR.bam \ -normal ${CONTROL} \ --germline-resource ${DB_PATH}/af-only-gnomad.raw.sites.hg19.vcf.gz \ --panel-of-normals ${NORMAL_BAM_DIR}/${CONTROL}.pon.vcf.gz \ --af-of-alleles-not-in-resource 0.0000025 \ --disable-read-filter MateOnSameContigOrNoMappedMateReadFilter \ --f1r2-tar-gz ${RESULT_DIR}/${sample_name}.f1r2.tar.gz \ -O ${RESULT_DIR}/${sample_name}.somatic.Mutect2.raw.vcf.gz -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 -I 분석할 BAM 또는 CRAM 파일을 지정 -tumor 분석할 종양 샘플의 ID를 지정 -normal (선택 사항) 분석할 정상 샘플의 ID를 지정 --disable-read-filter 기본적으로 활성화된 리드 필터 중 일부를 비활성화 --germline-resource 체세포 변이를 탐지할 때 참조할 알려진 생식세포 변이 (germline variant) 리소스 파일을 지정 --af-of-alleles-not-in-resource 리소스에 포함되지 않은 대립유전자의 가상 대립유전자 빈도(allele frequency)를 지정 --f1r2-tar-gz F1R2 통계 데이터를 저장할 파일을 지정(LearnReadOrientationModel 도구에 입력) -O 필터링된 변이 결과를 저장할 VCF 파일을 지정 -
Output
- ${sample_name}.somatic.Mutect2.raw.vcf.gz : 각 샘플별 somatic raw variant 파일
2) Calculate Contamination
Contamination을 제거하기 위하여 두 가지 단계를 진행합니다.
먼저, GetPileupSummaries 는 입력된 샘플의 BAM 파일에서 기존에 알려진 Germline VCF의 allele frequency (예. gnomAD VCF) 위치를 비교하여 ref_count, alt_count, allele_frequency 등을 계산하여 table을 구성합니다.
CalculateContamination 는 앞서 GetPileupSummaries 의 결과에서 확인된 Germline variant 정보를 바탕으로 cross-sample contamination이 발생할 수 있는 read 비율을 계산합니다.
FilterMutectCalls 는 CalculateContamination 을 통해 계산된 contam 으로 인해 발생한 variant 를 필터링합니다.
-
Input
- ${sample_name}.somatic.Mutect2.raw.vcf.gz : 각 샘플별 somatic raw variant 파일
-
Practice
### GetPileupSummaries ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ GetPileupSummaries \ -I ${TUMOR_BAM_DIR}/${sample_name}.sorted.RG.dedup.BQSR.bam \ -V ${DB_PATH}/small_exac_common_3.hg38.vcf.gz \ -O ${RESULT_DIR}/${sample_name}.tumor_getpileupsummaries.table ### CalculateContamination ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ CalculateContamination \ -I ${RESULT_DIR}/${sample_name}.tumor_getpileupsummaries.table \ -O ${RESULT_DIR}/${sample_name}.tumor_calculatecontamination.table ### FilterMutectCalls ### ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ FilterMutectCalls \ -V ${RESULT_DIR}/${sample_name}.somatic.Mutect2.raw.vcf.gz \ --contamination-table ${RESULT_DIR}/${sample_name}.tumor_calculatecontamination.table \ -O ${RESULT_DIR}/${sample_name}.somatic.Mutect2.contam_filtered.vcf.gz -
Option description
-V 필터링할 변이 호출이 포함된 VCF 파일을 지정 --contamination-table 샘플의 오염도를 나타내는 테이블 파일을 지정 -O 필터링된 변이 호출을 저장할 VCF 파일을 지정 -
Output
- ${sample_name}.somatic.Mutect2.contam_filtered.vcf.gz : contam 제거한 somatic vcf 파일
05-3. 한 개 이상의 샘플에 대한 변이 통합 분석
1) Consolidate GVCFs (GenomicDBImport) : 변이 결과 통합
하나의 샘플이 아닌 여러 개의 샘플에 대한 genotype을 한 번에 확인하기 위해 여러 샘플의 GVCF 파일을 통합하는 과정으로, GATK4 이전에는 CombineGVCFs 라는 방식으로 통합하였으나, 해당 방식은 파일을 생성하는 대신에 데이터 저장소를 생성하는 방식을 통해 성능이 높였습니다.
-
Input
- 여러 개의 GVCF 형태의 VCF 파일 예시 : HG002.raw.g.vcf.gz, HG003.raw.g.vcf.gz, HG004.raw.g.vcf.gz
-
Practice
### GenoicDBImport ### for j in {1..22} X Y M; ${GATK_PATH}/gatk \ --java-options -Xmx8g \ GenomicsDBImport \ -R ${REF_GENOME} \ -genomicsdb-workspace-path chr${j}_gdb \ --tmp-dir ${TEMP_DIR} \ --merge-input-intervals true \ --max-num-intervals-to-import-in-parallel 3 \ -L chr${j} \ --genomicsdb-shared-posixfs-optimizations true \ --batch-size 50 \ -V HG002.raw.g.vcf.gz \ -V HG003.raw.g.vcf.gz \ -V HG004.raw.g.vcf.gz; -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 --genomicsdb-workspace-path 통합된 gVCF 데이터베이스가 저장될 디렉터리 경로를 지정 --intervals / -L 데이터를 결합할 유전체 영역을 지정 --batch-size 한 번에 처리할 샘플의 최대 수를 지정 (Default : 50) --genomicsdb-shared-posixfs-optimizations 공유 POSIX 파일 시스템을 사용한 최적화를 활성화하는 옵션 --max-num-intervals-to-import-in-parallel 병렬로 동시에 가져올 최대 간격(유전체 구간) 수를 지정 --merge-input-intervals 지정된 간격이 서로 겹치는 경우 자동으로 병합하여 처리할지 여부를 지정 -
Output
- 1 ~ 22번, X, Y, M 에 해당하는 chr${j}_gdb 데이터베이스
2) Join-Call cohort (GenotypeGVCFs) : 샘플이 가진 변이만을 호출
특정 샘플의 GVCF 또는 많은 수의 샘플이 결합된 GVCF를 대상으로 샘플(들)이 가진 변이 정보만을 필터링하여 파일을 구성합니다. 이를 통해 필터링할 준비가 된 공동 호출 SNP 및 indel 호출 세트가 생성되며 모든 샘플에서 모든 관심 부위에 대한 정보를 제공합니다.
이 프로세스는 매우 빠르게 진행되며 특정 샘플이 코호트(N개)에 추가될 때 언제든지 다시 실행할 수 있으므로 소위 N+1 문제가 해결할 수 있습니다.
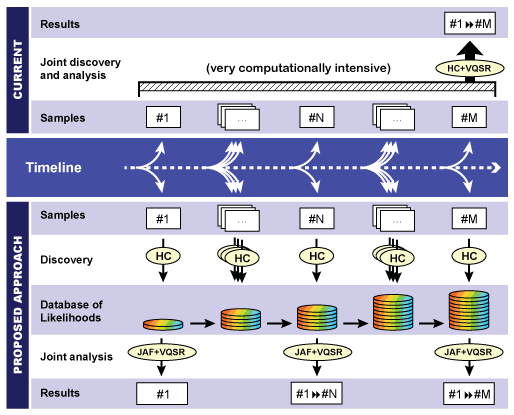
출처 : https://www.biostars.org/p/366846/
-
Input
- 1 ~ 22번, X, Y, M 에 해당하는 chr${j}_gdb 데이터베이스
-
Practice
${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ GenotypeGVCFs \ -R ${REF_GENOME} \ -V gendb://chr1_gdb \ -O ${RESULT_DIR}/chr1.vcf.gz ${GATK_PATH}/gatk --java-options "-Djava.io.tmpdir=/ldata/tmp" \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU" \ GenotypeGVCFs \ -R ${REF_GENOME} \ -V gendb://chr2_gdb \ -O ${RESULT_DIR}/chr2.vcf.gz ...(chr 3부터 22 진행 필요) #### merge vcf #### java -jar -Djava.io.tmpdir=/ldata/tmp \ -$XMX -XX:+UseParallelGC -XX:ParallelGCThreads=$NCPU ${PICARD_PATH}/picard.jar \ GatherVcfs \ I=chr1.vcf.gz I=chr2.vcf.gz I=chr3.vcf.gz I=chr4.vcf.gz I=chr5.vcf.gz I=chr6.vcf.gz I=chr7.vcf.gz I=chr8.vcf.gz I=chr9.vcf.gz I=chr10.vcf.gz I=chr11.vcf.gz I=chr12.vcf.gz I=chr13.vcf.gz I=chr14.vcf.gz I=chr15.vcf.gz I=chr16.vcf.gz I=chr17.vcf.gz I=chr18.vcf.gz I=chr19.vcf.gz I=chr20.vcf.gz I=chr21.vcf.gz I=chr22.vcf.gz I=chrX.vcf I=chrY.vcf.gz I=chrM.vcf.gz \ O=FAMILY.merged.vcf.gz -
Option description
-R 분석에 사용할 참조 유전체 파일을 지정 -V 입력으로 사용할 gVCF 파일이나 GenomicsDB 워크스페이스를 지정 -O 최종 호출된 변이 정보를 저장할 VCF 파일을 지정 -
Output
- FAMILY.merged.vcf.gz : chr을 모두 모아서 구성된 vcf 파일
3) Variant Quality Score Recalibration (VQSR) : 변이 품질 재조정
머신 러닝을 활용하여 훈련 세트의 변이 정보의 프로필을 모델링하고 이를 바탕으로 변이 호출에서 생성될 수 있는 artifact를 필터링하기 위하여 각 변이에 대한 품질 점수를 재조정하는 방식입니다.
핵심은 이미 잘 알려지고 검증된 변이 리소스(OMNI, 1000 Genomes, hapmap 등)를 사용하여 현재 변이 호출 결과에서 정답으로 확신할 수 있는 변이 집합과 그렇지 않은 집합을 선택한 후, 변이에 대한 정보를 기반으로 모델링하여 모든 변이에 적용하는 것입니다. 이를 통해 각 변이에 대한 품질점수를 산출할 수 있습니다. 즉 실제일 가능성이 높은 변이의 주석 프로필을 식별하고 호출자가 계산한 품질 점수(QUAL)보다 훨씬 더 신뢰할 수 있는 VQSLOD 점수를 각 변이에 할당합니다. 이 변이 품질 점수(VQSLOD)를 사용하여 원시 변이 세트를 필터링하고, 원하는 수준의 품질을 가진 변이들의 하위 세트를 생성하며, 특이성과 민감성의 균형을 맞추도록 미세 조정합니다.
-
Input
- FAMILY.merged.vcf.gz : chr을 모두 모아서 구성된 vcf 파일
-
Practice
${GATK_PATH}/gatk VariantRecalibrator \ -R ${REF_GENOME} \ -V ${WORKING_DIR}/FAMILY.merged.vcf.gz \ --resource:hapmap,known=false,training=true,truth=true,prior=15.0 hapmap_3.3.sites.vcf \ --resource:omni,known=false,training=true,truth=true,prior=12.0 1000G_omni2.5.sites.vcf \ --resource:1000G,known=false,training=true,truth=false,prior=10.0 1000G_phase1.snps.high_confidence.sites.vcf \ --resource:dbsnp,known=true,training=false,truth=false,prior=2.0 dbsnp_138.vcf \ -an DP -an QD -an MQ -an MQRankSum -an ReadPosRankSum -an FS -an SOR \ -mode SNP \ -O FAMILY.merged.cohort_SNP.recal \ --tranches-file FAMILY.merged.cohort_SNP.tranches ${GATK_PATH}/gatk ApplyVQSR \ -R ${REF_GENOME} \ -V ${WORKING_DIR}/FAMILY.merged.vcf.gz \ --truth-sensitivity-filter-level 99.0 \ --tranches-file FAMILY.merged.cohort_SNP.tranches \ --recal-file FAMILY.merged.cohort_SNP.recal \ -mode SNP \ -O FAMILY.merged.cohort.genotyped.VQSR.snp.vcf -
Option description
-V 변이 정보를 포함한 VCF 파일을 지정 -R 분석에 사용할 참조 유전체 파일을 지정 --mode 재조정할 변이의 종류를 지정 SNP SNP(단일 염기 다형성)를 재조정 INDEL InDel(삽입 및 결실)을 재조정 BOTH SNP와 InDel을 동시에 재조정 --resource 모델 학습에 사용할 외부 변이 데이터(리소스)를 지정 -
Output
- FAMILY.merged.cohort.genotyped.VQSR.snp.vcf : 해당 cohort에서의 SNP 수행결과
-
Recalibration model을 만들기 위한 resource
- 참고 링크 : https://github.com/broadgsa/gatk/blob/master/doc_archive/tutorials/(howto)_Recalibrate_variant_quality_scores_=_run_VQSR.md
-
HapMap (truth=true, training=true)
- SNP call set that has been validated to a very high degree of confidence
-
Omni (truth=true, training=true)
- a set of polymorphic SNP sites produced by the Omni genotyping array
-
1000G (truth=false, training=true)
- a set of high-confidence SNP sites produced by the 1000 Genomes Project
-
dbSNP (truth=false, training=false, known=true)
- a SNP call set that has not been validated to a high degree of confidence
- 이 데이터 세트를 통해 dbSNP에 변이가 있는지 여부를 판단합니다
06. Annotate variants
Germline, Somatic Variant calling을 통해 획득한 변이(variant)에 대한 정보를 파악하기 위하여 refGene, 1000genome, dbnsfp 등의 잘 알려진 변이 데이터베이스를 통해 각 변이들에 대한 주석 정보를 추가하는 것입니다. 이를 통해 pathogenic, benign 한 변이들을 구분할 수 있어 질병에 원인이 되는 변이를 추출할 수 있습니다.
06-1. ANNOVAR Annotation
ANNOVAR : https://annovar.openbioinformatics.org/en/latest/
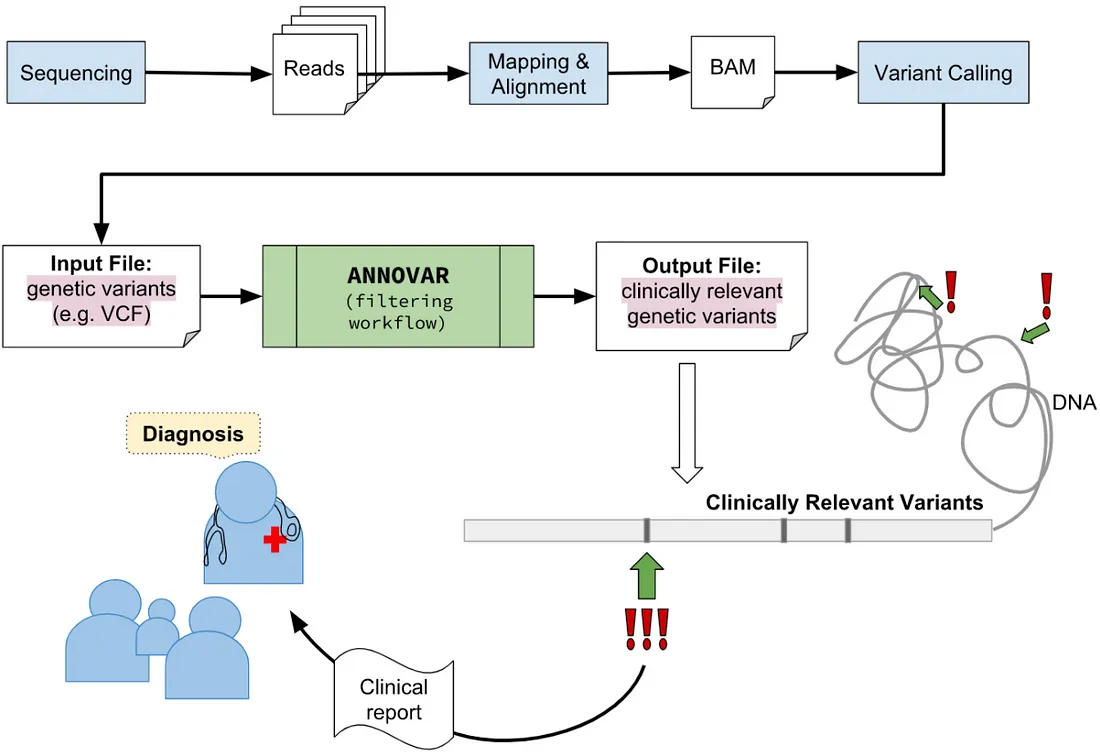
ANNOVAR 는 다양한 유전체(인간 hg18, hg19, hg38, hs1(T2T-CHM13) 및 마우스, 벌레, 파리, 효모 등)에서 검출된 유전적 변이에 기능적으로 주석을 달기 위해 최신 정보를 활용하는 효율적인 프로그램으로 변이에 대한 Chr, start position, end position, ref allele, alt allele 등의 정보를 통해 분석을 수행할 수 있습니다.
1) convert2annovar.pl 을 통한 annovar input 파일로 변환
1. VCF 파일에 있는 변이를 ANNOVAR이 이해할 수 있는 간단한 텍스트 형식으로 변환 합니다. 이 변환된 파일은 이후 ANNOVAR 주석을 실행할 때 입력으로 사용됩니다.
2) table_annovar.pl 을 통한 annotation 수행 방법
1. gene-based : SNP 또는 CNV가 단백질 코딩 변화를 일으키는지 여부와 영향을 받는 아미노산을 식별하여 주석 정보를 추가합니다 (RefSeq 유전자, UCSC 유전자, ENSEMBL 유전자, GENCODE 유전자 등을 활용합니다)
2. position-based : 44개 종 간의 conserved region, 예측 전사 인자 결합 부위, CNV 영역, GWAS 정보, 변이 데이터베이스, DNAse I, ENCODE 의 히스톤 정보, ChIP-Seq peak 등 많은 주석 등 특정 게놈 영역의 변이를 식별할 수 있습니다.
3. filter-based : 특정 데이터베이스에 기록된 변이를 식별하여 대립 유전자의 빈도 혹은 단백질 계수( SIFT/PolyPhen/FATHMM/MetaSVM/MetaLR 점수)를 통해 변이에 대한 주석을 추가할 수 있습니다.
-
Input
- germline, somatic variant vcf 파일 : HG002.snp.PASS.vcf
-
Practice
# 1) convert2annovar.pl 을 통한 annovar input 파일로 변환 ### convert2annovar input ### ${ANNOVAR_PATH}/convert2annovar.pl \ HG002.snp.PASS.vcf \ -format vcf4 \ > HG002.snp.PASS.step1 # 2) table_annovar.pl 을 통한 Annotation 수행 ### MAF >= 0.1, 1000Genome, polyphen ### ${ANNOVAR_PATH}table_annovar.pl \ HG002.snp.PASS.step1 \ -buildver hg38 \ -remove \ -out HG002 \ -protocol refGene,1000g2015aug_all,dbnsfp35a,exac03,gnomad_genome,gnomad_exome,esp6500siv2_all \ -operation g,f,f,f,f,f,f -
Option description
-out 생성할 출력 파일의 이름을 지정 -protocol 사용할 주석 데이터베이스를 지정 -operation 각 데이터베이스에 대해 수행할 작업 유형을 지정 -remove 분석 완료 후 중간 산출물 삭제 -buildver 특정 유전체 빌드 버전을 명시 -
Output
- HG002.hg38_multianno.txt : 각 변이에 대해 annotation 된 텍스트 파일