Guides
- Tools for Alignment workflow
- Reference Genome
- DNA sequencing pipeline command-line parameters
- Tools for Somatic Mutation call workflow
- Somatic Mutation call pipeline command-line parameters
- Tools for Methylation call workflow
- Reference Genome
- Methylation sequencing pipeline command-line parameters
DNA Sequencing Analysis Guide
Workflow summary

| Name | Type | Version |
|---|---|---|
| Reference genome | File | GRCh38.p14 |
| Germline resource | File | af-only-gnomad.vcf.gz |
| Panel of normals | File | pon.vcf.gz |
| dbSNP | File | dbsnp155 |
| BWA-Mem | Software | 0.7.17 |
| GATK (Mutect2) | Software | 4.4.0.0 |
| Samtools | Software | 1.18 |
| MuSE | Software | 2.0.3 |
| VarScan2 | Software | 2.4.6 |
| Pindel | Software | 0.2.5b9 |
| VEP | Software | 111 |
Tools for Alignment workflowTools for Alignment workflow
The BWA-MEM algorithm is used in the map/alignment step, and directly converted into a bam file using Samtools. Tumor and normal samples are labeled separately in read groups, and sorted by chromosome using GATK-Picard’s Sortsam. Duplication marks and base recalibration are then performed using GATK. See below for detailed command-line scripts.
DNA Sequencing Analysis GuideReference Genome
All sequence reads were aligned using the human reference genome hg38 which is the UCSC modified version of GRCh38.p14 released in June 2022. More information is here.
DNA sequencing pipeline command-line parameters
Step 1 : Map/Align (BWA-MEM)
bwa mem -t <Num threads> \
-R <read group> \
<reference fasta> \
<R1 fastq> <R2 fastq> | \
samtools view -Shb –o <out bam>
Step 2 : Sort bam (GATK-Picard)
gatk SortSam \
-I <in bam> \
-O <out bam> \
-SO coordinate \
--VALIDATION_STRINGENCY SILENT \
--MAX_RECORDS_IN_RAM 50000000 && \
samtools index <out bam>
Step 3 : Mark duplicate (GATK)
gatk MarkDuplicates \
-I <in bam> \
-O <marked_dup bam> \
-M <marked_dup metrics> \
--VALIDATION_STRINGENCY SILENT \
--MAX_RECORDS_IN_RAM 50000000 && \
samtools index <marked_dup bam>
Step 4 : Recalibrate Base Quality Scores (GATK)
gatk BaseRecalibrator \
-R <reference fasta> \
-I <in bam> \
--known-sites <known sites vcf> \
-O <recal table>
gatk ApplyBQSR \
-R <reference fasta> \
-I <in bam> \
--bqsr-recal-file <recal table> \
-O <recalibrated bam> \
--create-output-bam-index false && \
samtools index <recalibrated bam>
Tools for Somatic Mutation call workflow
The map/aligned bam files are analyzed for somatic mutations using four tools: Mutect2, MuSE, VarScan2, and Pindel. Mutation calls are reported by each tool in a VCF file.
Somatic Mutation call pipeline command-line parameters
Mutect2 (GATK) Germline resource: af-only-gnomad.vcf.gz Panel of normals: pon.vcf.gz
gatk Mutect2 \
-R <reference fasta> \
-I <tumor bam> \
-I <normal bam> \
-normal normal \
--germline-resource <af-only-gnomad.vcf.gz> \
--panel-of-normals <pon.vcf.gz> \
-O <out vcf>
MuSE dbSNP: dbsnp155
# MuSE call Step
MuSE call \
<Tumor bam> <Normal bam>\
-f <reference fasta>\
-O <muse call intermediate>
# MuSE sump Step
MuSE sump \
-I <muse call intermediate>\
-G \ # -G and -E stand for WGS and WES, respectively
-D <dbsnp155.vcf.gz>\
-O <out vcf>
VarScan2
# Mpileup Step via samtools
-f <reference fasta> \
-q 0 \
-B \
<Normal bam> <Tumor bam> \
<intermediate_mpileup.pileup>
# Somatic mutation call
varscan somatic \
<intermediate_mpileup.pileup> \
<out prefix> \
--mpileup 0 \
--min-coverage 8 \
--min-coverage-normal 8 \
--min-coverage-tumor 6 \
--min-var-freq 0.10 \
--min-freq-for-hom 0.75 \
--normal-purity 1.0 \
--tumor-purity 1.00 \
--p-value 0.99 \
--somatic-p-value 0.05 \
--strand-filter 0 \
--output-vcf 1
# Mutation classification
varscan processSomatic \
<intermediate vcf> \
--min-tumor-freq 0.10 \
--max-normal-freq 0.05 \
--p-value 0.07
Pindel
sambamba view <Tumor bam> \
--filter "\not (unmapped or duplicate or secondary_alignment or failed_quality_control or supplementary)\" \
--format bam \
--output-filename <Tumor sambamba bam> \
gatk CollectInsertSizeMetrics \
-I <Tumor sambamba bam> \
-O <Tumor_insert_size_metric.txt> \
-H <Tumor_insert_size_metric.pdf> && \
sambamba view <Normal bam> \
--filter "\not (unmapped or duplicate or secondary_alignment or failed_quality_control or supplementary)\" \
--format bam \
--output-filename <Normal sambamba bam> \
gatk CollectInsertSizeMetrics \
-I <Normal sambamba bam> \
-O <Normal_insert_size_metric.txt> \
-H <Normal_insert_size_metric.pdf> && \
t_size=$(printf "%.0f" "$(head -8 <Tumor_insert_size_metric.txt> | tail -1 | awk 'BEGIN{FS=OFS="\t";}{print $6;}')") \
n_size=$(printf "%.0f" "$(head -8 <Normal_insert_size_metric.txt> | tail -1 | awk 'BEGIN{FS=OFS="\t";}{print $6;}')") \
echo -e "<Tumor sambamba bam>\t\ttumor" > <Tumor>_<Normal>.pindel.config \
echo -e "<Normal sambamba bam>\t\tnormal" >> <Tumor>_<Normal>.pindel.config && \
pindel \
-f <reference fasta> \
-i <Tumor>_<Normal>.pindel.config \
-o <Output prefix> && \
grep ChrID <Output prefix>.D > <Output prefix>.D.head \
grep ChrID <Output prefix>.SI > <Output prefix>.SI.head && \
cat <Output prefix>.D.head <Output prefix>.SI.head > <Output prefix>.pindel_somatic && \
cat << EOF > <Output prefix>.somatic.indel.filter.config
indel.filter.input = <Output prefix>.pindel_somatic
indel.filter.vaf = 0.08
indel.filter.cov = 20
indel.filter.hom = 6
indel.filter.pindelvcf = <PINDEL path>
indel.filter.reference = <reference fasta>
indel.filter.referenceindex = <genome build>
indel.filter.referencedate = $(date '+%Y%m%d')
indel.filter.output = <Output prefix>_filter.vcf
EOF && \
perl <PINDEL path>/bin/somatic_filter/somatic_indelfilter.pl <Output prefix>.somatic.indel.filter.config && \
gatk SortVcf \
-I <Output prefix>_filter.vcf \
-O <Output prefix>_filter_sorted.vcf \
-SD <reference dictionary file> \
--CREATE_INDEX false && \
tabix -p vcf <Output prefix>_filter_sorted.vcf.gz && \
gatk VariantFiltration \
-R <Reference fasta> \
-V <Output prefix>_filter_sorted.vcf.gz \
-O <Output prefix>_filtered.vcf.gz \
--filter-name TALIDP \
--filter-expression "\(vc.isbiallelic() && vc.getGenotype(\"TUMOR\").getAD().1 < 3\)" \
--create-output-variant-index false && \
tabix -p vcf <Output prefix>_filtered.vcf.gz
Mutation annotation command line parameters vep cache : Ensembl v.111
vcf2maf.pl --input-vcf <Tumor-Normal vcf> \
--output-maf /root/output/$2_varscan.hc.maf \
--tumor-id <tumor sample ID> \
--normal-id <Normal sample ID> \
--vcf-tumor-id TUMOR \
--vcf-normal-id NORMAL \
--ref-fasta <reference fasta> \
--cache-version 111 \
--ncbi-build GRCh38
RNA Sequencing Analysis Guide
| Name | Type | Version |
|---|---|---|
| Reference genome | File | GRCh38.p14 |
| GENCODE | File | v44 |
| Ensembl | File | v110 |
| refGene | File |
Hg38 |
| STAR | Software | 2.7.11a |
| RSEM | Software | 1.3.3 |
| STAR-Fusion | Software | 1.13.0 |
Tools for Alignment workflow
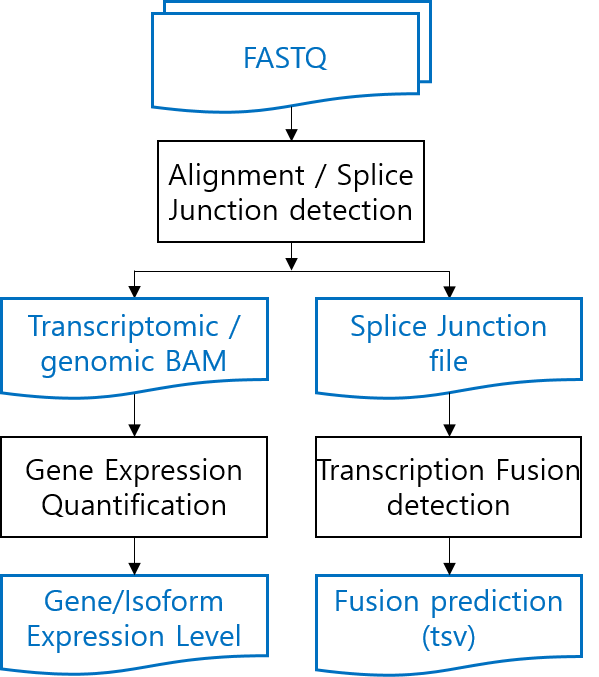
The alignment step uses a STAR's two-pass method, which includes the detection of new splice junctions. In the alignment step, STAR generates a transcriptomic and a genomic BAM, from which the gene expression is calculated using the rsem-calculate-expression, sub-command of RSEM. And in the junction detection process, STAR generates a tab-delimited file containing the splice junction information, from which the fusion gene information is predicted using STAR-Fusion. See below for detailed command-line scripts.
Reference Genome
All sequence reads were aligned using the human reference genome hg38 which is the UCSC modified version of GRCh38.p14 released in June 2022 More information is here. Gene models use GENCODE v44, Ensembl v110, and refGene models.
RNA sequencing pipeline command-line parameters
Step 1 : Alignment/Splice Junction detection (STAR)
STAR \
--readFilesIn <R1 fastq> <R2 fastq> \
--outSAMattrRGline ID:<sample name> \
--genomeDir <reference path> \
--readFilesCommand zcat \
--twopassMode Basic \
--outFilterMultimapNmax 20 \
--alignSJoverhangMin 8 \
--alignSJDBoverhangMin 1 \
--outFilterMismatchNmax 999 \
--outFilterMismatchNoverLmax 0.1 \
--alignIntronMin 20 \
--alignIntronMax 1000000 \
--alignMatesGapMax 1000000 \
--outFilterType BySJout \
--outFilterScoreMinOverLread 0.33 \
--outFilterMatchNminOverLread 0.33 \
--limitSjdbInsertNsj 1200000 \
--outFileNamePrefix / <output path> / <sample name>. \
--outSAMstrandField intronMotif \
--outFilterIntronMotifs None \
--alignSoftClipAtReferenceEnds Yes \
--quantMode TranscriptomeSAM GeneCounts \
--outSAMtype BAM SortedByCoordinate Unsorted \
--outSAMunmapped Within \
--genomeLoad NoSharedMemory \
--chimSegmentMin 15 \
--chimJunctionOverhangMin 15 \
--chimOutType Junctions SeparateSAMold WithinBAM SoftClip \
--chimOutJunctionFormat 1 \
--chimMainSegmentMultNmax 1 \
--outSAMattributes NH HI AS nM NM ch
Step 2 : Gene Expression Quantification (RSEM)
rsem-calculate-expression \
--no-bam-output \
--forward-prob 0 \
--estimate-rspd \
--alignments \
--paired-end \
<in bam> \
<refenence fasta> \
<output prefix>
Step 3 : Transcription Fusion Detection (STAR-Fusion)
STAR-Fusion \
--genome_lib_dir <genome lib dir> \
-J <in file> \
--output_dir <output path>
Methylation Sequencing Analysis Guide

| Name | Type | Version |
|---|---|---|
| Reference genome | File | GRCh38.p14 |
| Bowtie2 | Software | 2.5.2 |
| Bismark | Software | 0.24.2 |
Tools for Methylation call workflow
The alignment step used the Bowtie2 and HISAT2 algorithms, and then removed duplicate alignments at the same location in the genome from the alignment results. The methylation extraction step created output for each of the three different sequence contexts (CpG, CHG or CHH) and generated a bedgraph formatted file using the bedgraph option. All of this analysis was done through the Bismark pipeline. See below for detailed command-line scripts.
Reference Genome
All sequence reads were aligned using the human reference genome hg38 which is the UCSC modified version of GRCh38.p14 released in June 2022. More information is here.
Methylation sequencing pipeline command-line parameters
Step 1 : Alignment
bismark \
--genome <reference path> \
-1 <R1 fastq> \
-2 <R2 fastq> \
-o <output path>
Step 2 : Deduplication
deduplicate_bismark \
--bam <input bam> \
-o <output bam> \
--output_dir <output path>
Step 3 : Methylation calling
bismark_methylation_extractor \
--gzip \
--bedGraph \
<in bam> \
--output <output path>
Methylation Array Analysis Guide
| Name | Type | Version |
|---|---|---|
| minfi (R package) | Software | 1.50.0 |
| ChAMP (R package) | Software | 2.29.1 |
Tools for Methylation array workflow
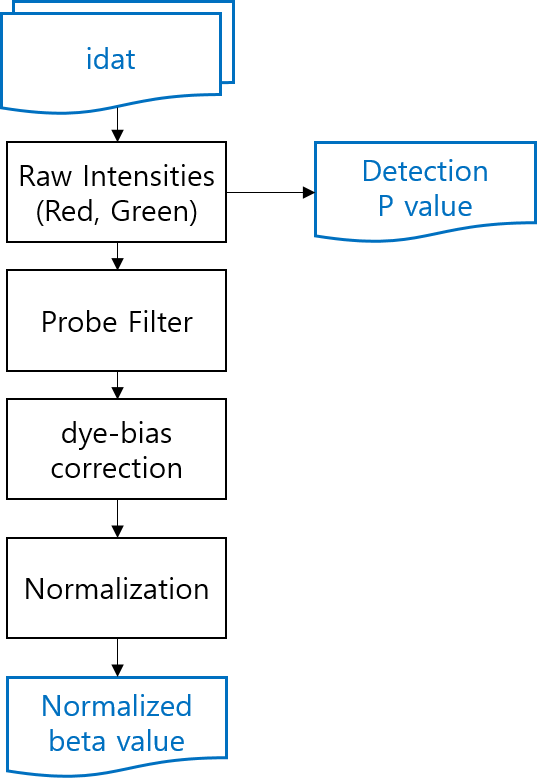
The methylation array analysis basically follows the minfi protocol (R package). First, idat files were converted to RGChannelSet, which contains the raw intensities in the green and red channels. Probes below a certain threshold were removed using the detection p-value. Normalization was a two-step normalization using the Noob method followed by the BMIQ method. Background correction (dye bias correction) was performed in the Noob method and normzlisation was performed in the Noob and BMIQ methods. The majority of the process was performed using minfi and the BMIQ normalization was performed using ChAMP. See below for detailed command-line scripts.
Reference Genome
See minfi and ChAMP bioconductor page.
Methylation array pipeline command-line parameters
Step 1 : data import and convert into RGChannelSet
basedir ‘/BASEDIR'
targets read.metharray.sheet(basedir, pattern = "csv$", ignore.case = TRUE, recursive = TRUE, verbose = TRUE)
rgset read.metharray.exp(targets = targets, force = TRUE, extended = TRUE)
Step 2 : Extraction of detection p value
det_p detectionP(rgset, type = "m+u")
Step 3 : Normalization (2-way)
# Noob method
norm_by_noob preprocessNoob(rgset, offset = 15, dyeCorr = TRUE, verbose = TRUE, dyeMethod = c("single"))
# BMIQ method
norm_by_bmiq champ.norm(beta = getBeta(norm_by_noob), mset = norm_by_noob, method = "BMIQ", cores = 1, arraytype = platform, plotBMIQ = FALSE)